

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website https://www.jofem.org |
Case Report
Volume 10, Number 5, October 2020, pages 144-153
Adrenal Insufficiency due to Total Primary Empty Sella Syndrome
Daisuke Usudaa, b, e, Susumu Takagic, Kohei Takanagaa, Toshihide Izumidaa, Ryusho Sangena, Toshihiro Higashikawad, Yuji Kasamakia
aDepartment of General Medicine, Kanazawa Medical University Himi Municipal Hospital, Himi-shi, Toyama-ken 935-8531, Japan
bDepartment of Infectious Diseases, Kanazawa Medical University, Uchinada-machi, Ishikawa-ken 920-0293, Japan
cDepartment of Diabetology and Endocrinology, Kanazawa Medical University Himi Municipal Hospital, Himi-shi, Toyama-ken 935-8531, Japan
dDepartment of Geriatric Medicine, Kanazawa Medical University Himi Municipal Hospital, Himi-shi, Toyama-ken 935-8531, Japan
eCorresponding Author: Daisuke Usuda, Department of General Medicine, Kanazawa Medical University Himi Municipal Hospital, 1130 Kurakawa, Himi-shi, Toyama-ken 935-8531, Japan
Manuscript submitted August 19, 2020, accepted August 26, 2020, published online October 16, 2020
Short title: Adrenal Insufficiency by ESS
doi: https://doi.org/10.14740/jem686
| Abstract | ▴Top |
A 64-year-old woman was transported suffering from persistent lower abdominal pain, vomiting, and low-grade fever. Magnetic resonance imaging revealed an empty sella (ES) and hormone tests revealed a disappearance of diurnal variation of cortisol, low cortisol and adrenocorticotropic hormone (ACTH) secretion especially in the morning, and poor ACTH-cortisol axis reaction, as well as normal hypothalamus-pituitary gland-thyroid or adrenal gland axis hormone reaction. The cause of ES remained unclear; however, based on a diagnosis as adrenal insufficiency due to inappropriate ACTH secretion caused by total primary ES syndrome, we started hydrocortisone (15 mg/day). Afterwards, she immediately became symptom-free and was discharged.
Keywords: Adrenal insufficiency; Cortisol; Adrenocorticotropic hormone; Empty sella syndrome
| Introduction | ▴Top |
“Empty sella” (ES) refers to the neuroradiological or pathological finding of an apparently ES turcica that contains no pituitary tissue [1]. An ES develops when cerebrospinal fluid (CSF) fills the sella turcica, compressing pituitary tissue until it lines the floor and walls of the sella, and when there is remodeling of the sella turcica and a flattening of the pituitary gland that results from subarachnoid space extension into an intra-seller position and a stretching of the pituitary stalk [2-6]. ES syndrome (ESS) refers to an anatomical and radiological condition, first described by Busch in 1951 [6]. ESS is complete or partial, depending on whether the sella turcica is completely or partially filled with CSF; this results in displacement of the pituitary gland, and therefore ESS is the pathological variant [6, 7]. In partial ESS, there is a pituitary gland thickness of 3 - 7 mm, with the sella filled less than 50% with CSF [6]. On the other hand, total ESS refers to when the pituitary gland thickness is less than 2 mm, and spinal fluid fills over half of the sella [6]. ESS patients have one or more pituitary hormone deficiencies [6].
Regarding pathophysiology and etiology, ESS is subdivided into two categories: primary ESS (PES) and secondary ESS (SES). PES occurs when there is increased CSF pressure alongside a defect in the diaphragma sellae [2, 3, 6, 8-10]. While there is no clear genetic association known to cause a predisposition to PES, it is likely that the incompetent diaphragma sellae was present at the patient’s birth [3]. The pathogenetic mechanisms of PES are not well established, but an ischemic atrophy of the adenohypophysis may be involved in the development of a PES with idiopathic chronic raised intracranial pressure, preventing the recovery of the gland volume after the intracranial pressure is restored to normal values [8, 11]. Restitution of ES may also be an indicator of ordinary intracranial pressure [11]. This is rarely known to cause pituitary dysfunction [6, 12]. On the other hand, there is a reported case in which a diagnosis of PES with anterior pituitary dysfunction was made in the absence of any history of pituitary irradiation, pituitary adenoma, or surgery [6]. SES can occur as a result of damage to the pituitary itself (e.g., pituitary apoplexy) or as a consequence of surgery, radiation treatment, immunotherapy or other novel forms of treatment, hemorrhage, autoimmune hypophysitis, neurosarcoidosis, or an infarction of the pituitary gland, and it may occur at any time during the patient’s life [2, 3, 6, 13-16]. In addition, a relationship has been reported between infection with Hantaan virus and SES [17].
In adults, ES is most commonly found in older women who have given birth to multiple children, and who are obese and suffer from hypertension, and it may be asymptomatic [2, 6, 18]. In children, however, ES is more frequently associated with clinical symptoms and endocrinopathies, particularly growth hormone (GH) deficiency, hypogonadotropism, or multiple pituitary hormone deficiencies [2]. Regardless of body mass index, PES patients show an increased cardiovascular [12]. In addition, there is an association between worse lipid and glucose profiles, and higher Framingham scores, with secondary hypothyroidism, even subclinical, as well as hypogonadism [12].
As far as we are aware, we have reported the first known case of adrenal insufficiency due to insufficient adrenocorticotropic hormone (ACTH) secretion to be caused by total PES, together with a brief review of literature. This case would help clinicians for procedure of diagnosis and treatment for ESS.
| Case Report | ▴Top |
A 64-year-old woman was transported suffering from persistent lower abdominal pain, vomiting, and low-grade fever. She had also been transported to our hospital 2 years ago due to the same complaint. Her medical history included total blindness when she was in her 30s, undergoing fibroid surgery when she was in her 40s, schizophrenia, type 2 diabetes mellitus without any complications, hypertension, and dyslipidemia. She was treated with risperidone, metformin, voglibose, candesartan, and pravastatin. The patient was unemployed and did not have any food or drug allergies. She had never experienced abnormal menstruation, including amenorrhea, and had a son. She had no family history of any immunodeficiency disorders or other congenital anomalies. She was independent for everyday activities, lived alone, and received food delivery services. On the other hand, she had sometimes complained about fatigue or abdominal pain, especially in the morning, according to her detailed history. This case report was approved by the Kanazawa Medical University Himi Municipal Hospital ethics committee and carried out in conformance with the principles of the Declaration of Helsinki.
She was 150 cm tall and weighed 57 kg, and she was obese, but without moon face, skin rashes, or striae. Her vital signs were abnormal, with a blood pressure of 104/58 mm Hg, a heart rate of 76 regular beats/min, a body temperature of 37.6 °C, oxygen saturation of 97% in ambient air, and a respiratory rate of 16/min; her Glasgow Coma Scale score was 15 (Eye (E) 4 Verbal (V) 5 Motor (M) 6) points. She complained of tenderness throughout the entire abdomen, and nothing else abnormal, including skin findings, was detected upon physical examination. A routine laboratory examination revealed increased values of white blood cells, lactate dehydrogenase, creatine kinase, and decreased values of blood urea nitrogen, sodium, and chloride. On the other hand, other values were normal, including complete blood count, biochemistry, casual blood glucose, ammonia, and urine tests (Table 1). In addition, no dramatic hormone secretion abnormality was confirmed in spotting pituitary, thyroid, and adrenal gland hormone examinations. In an imaging examination, a cranial computed tomography (CT) scan revealed ES in the pituitary gland with a height of 1 mm (Fig. 1). A magnetic resonance imaging (MRI) scan found CSF filling the sella turcica, and its intensity was the same as the cerebral ventricle (iso-intensity in T1-weighted image, and high intensity in T2-weighted image), in addition to the similar findings of the CT scan (Fig. 2a, b). On the other hand, chest and abdominal CT scans revealed normal findings. At this point, we suspected her diagnosis was ESS, which led to insufficient ACTH secretion and adrenal insufficiency. After she was hospitalized, we measured daily ACTH and cortisol secretion (Fig. 3) and performed two hormone load tests, namely an insulin tolerance test (ITT) and intravenous (IV) administration of Novolin R® 6 U (0.1 U/kg), and an anterior pituitary function test through combined IV administration of four hypothalamic releasing hormones: corticotropin-releasing hormone (CRH), thyrotropin-releasing hormone (TRH), luteinizing hormone-releasing hormone (LH-RH), and growth-hormone releasing factor (GRF) (Figs. 4, 5). As a result, we confirmed a disappearance of diurnal variation of cortisol, low ACTH secretion in the morning, and poor ACTH-cortisol axis reaction as well as normal hypothalamus-pituitary-gland-thyroid or adrenal gland axis hormone reaction. More specifically, on the ITT, a hypoglycemic state occurred 30 min after insulin administration and continued until 90 min after; on the other hand, we could not regard this as single ACTH deficiency, because its secretion was confirmed. Other pituitary hormones had been secreted adequately by the hypothalamus upon release of hormone stimulation. On the other hand, her daily cortisol secretion to urine was 58.8 mg/dL/day and was within normal range.
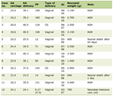 Click to view | Table 1. Result of Laboratory and Urine Examination |
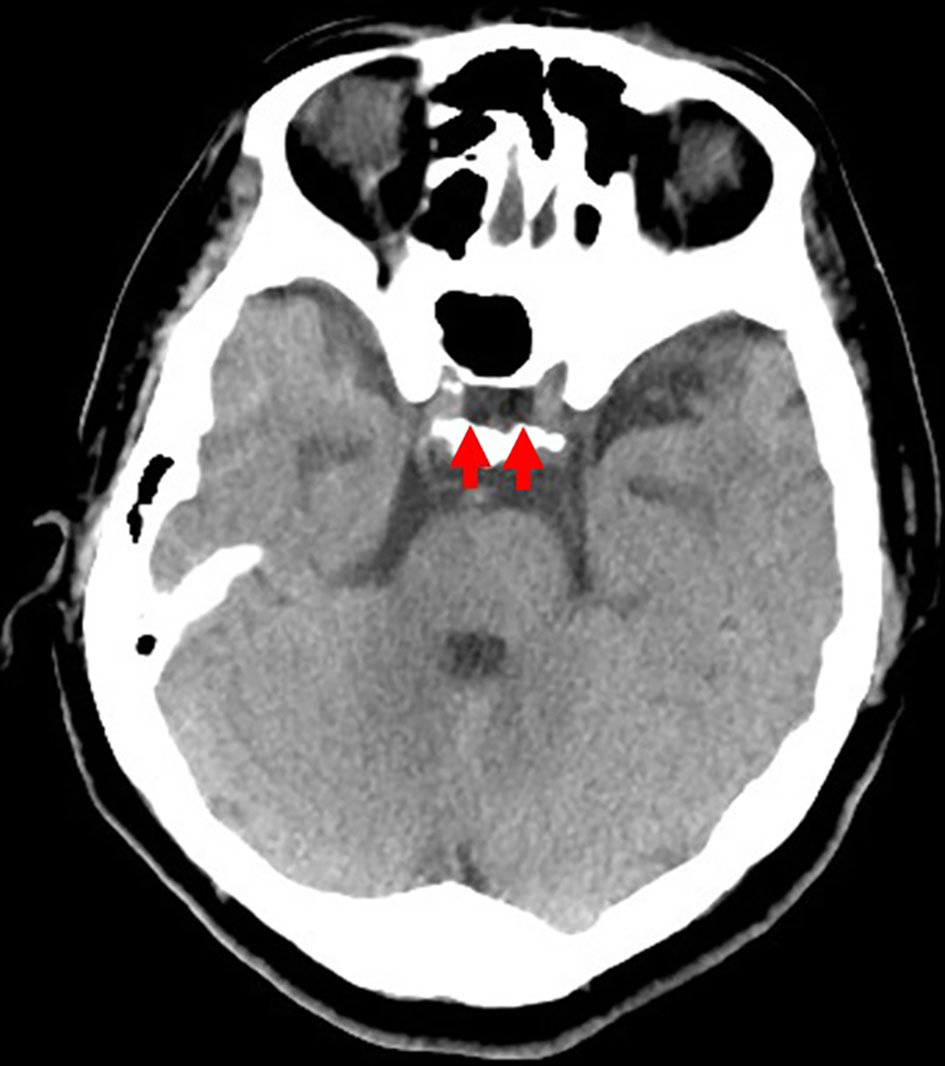 Click for large image | Figure 1. Cranial computed tomography scan from the emergency room. An empty sella is confirmed in the pituitary gland (red arrow), with a height of 1 mm. |
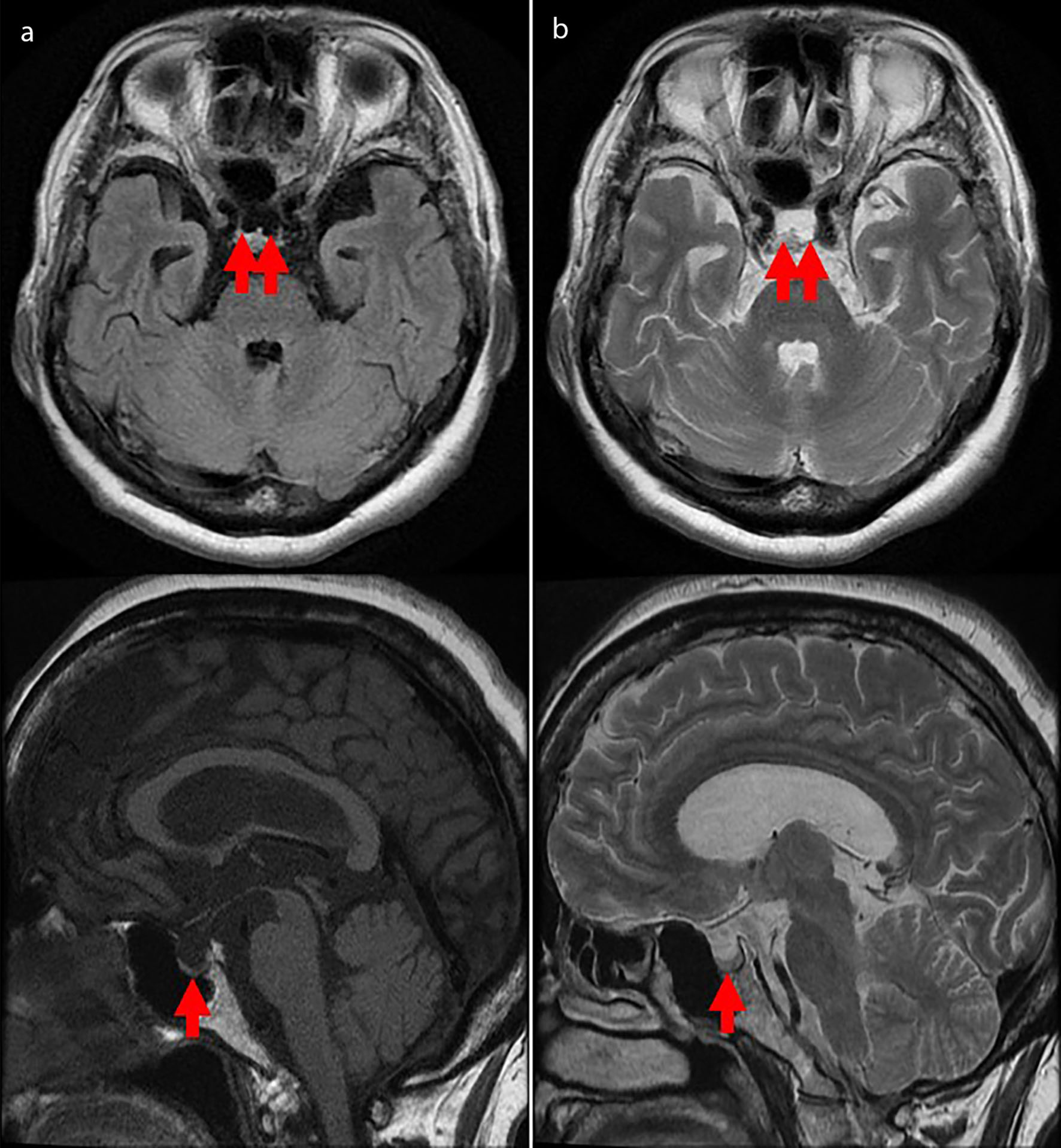 Click for large image | Figure 2. Cranial magnetic resonance imaging scan from the emergency room. An empty sella is confirmed in the pituitary gland (red arrow), with a height of 1 mm. Cerebrospinal fluid has filled the sella turcica and its intensity is the same as the cerebral ventricle (iso-intensity in T1-weighted image (a), high intensity in T2-weighted image (b)). |
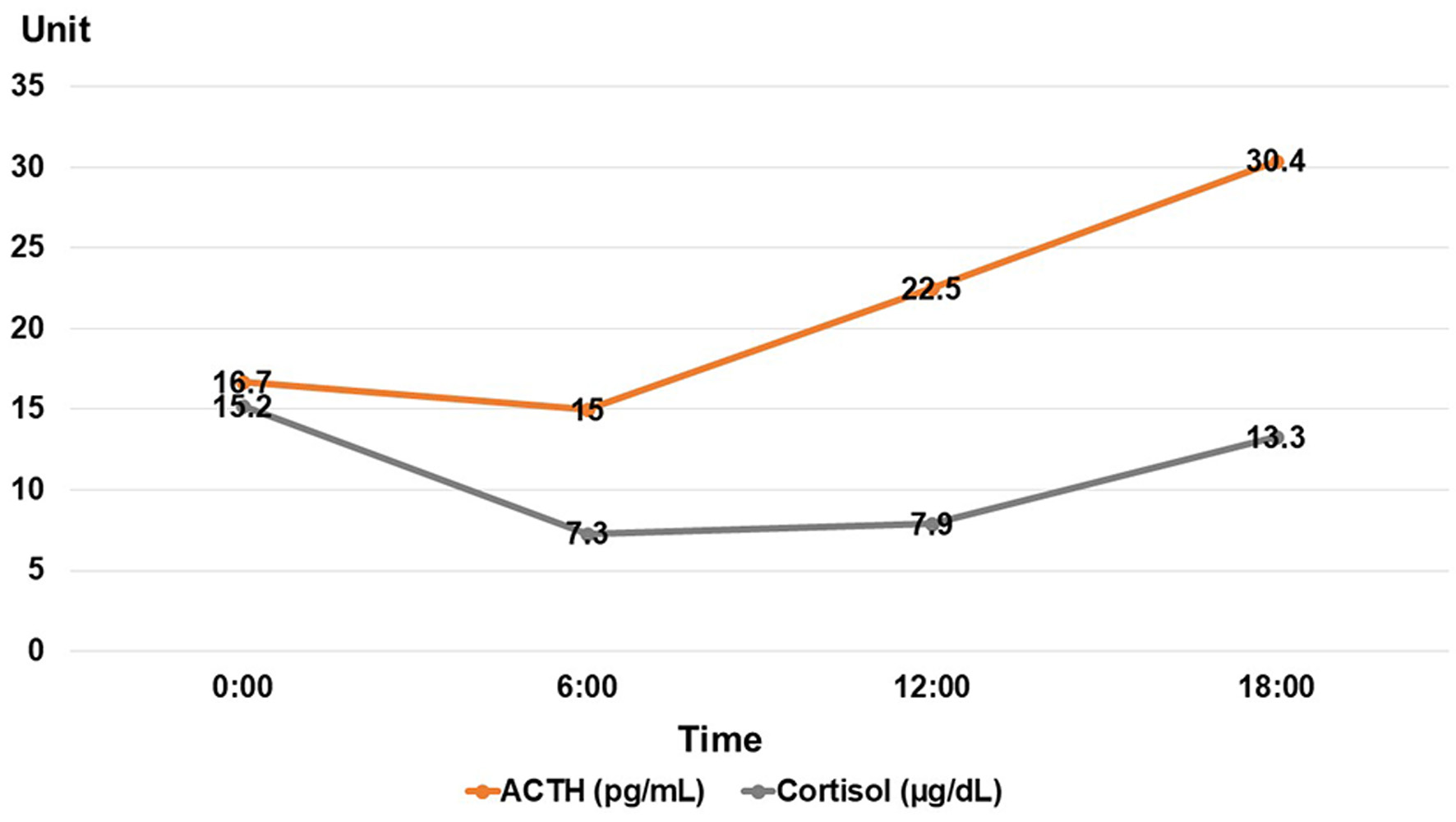 Click for large image | Figure 3. Daily ACTH and cortisol secretions. ACTH: adrenocorticotropic hormone. |
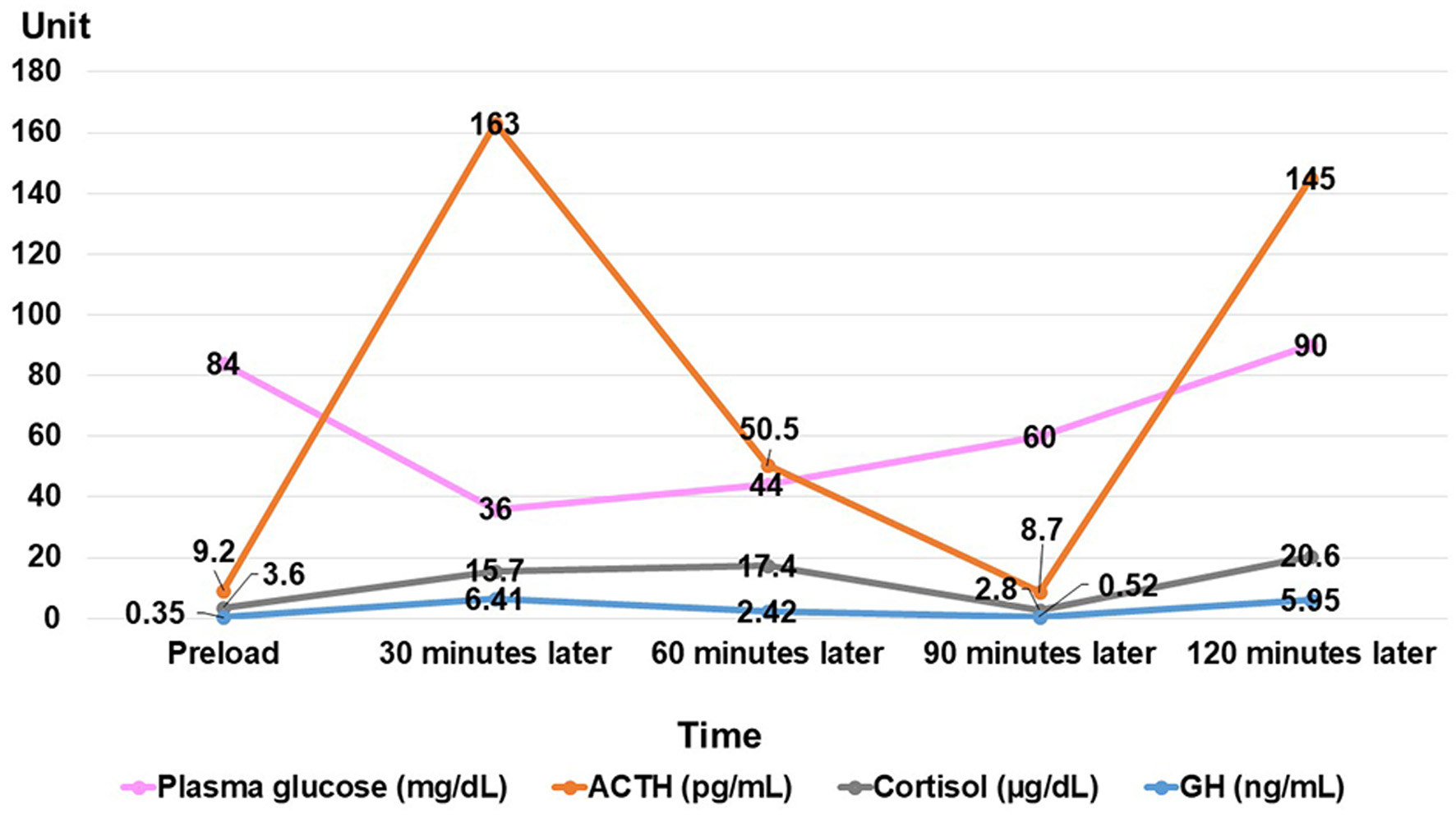 Click for large image | Figure 4. Result of insulin tolerance test. ACTH: adrenocorticotropic hormone; GH: growth hormone. |
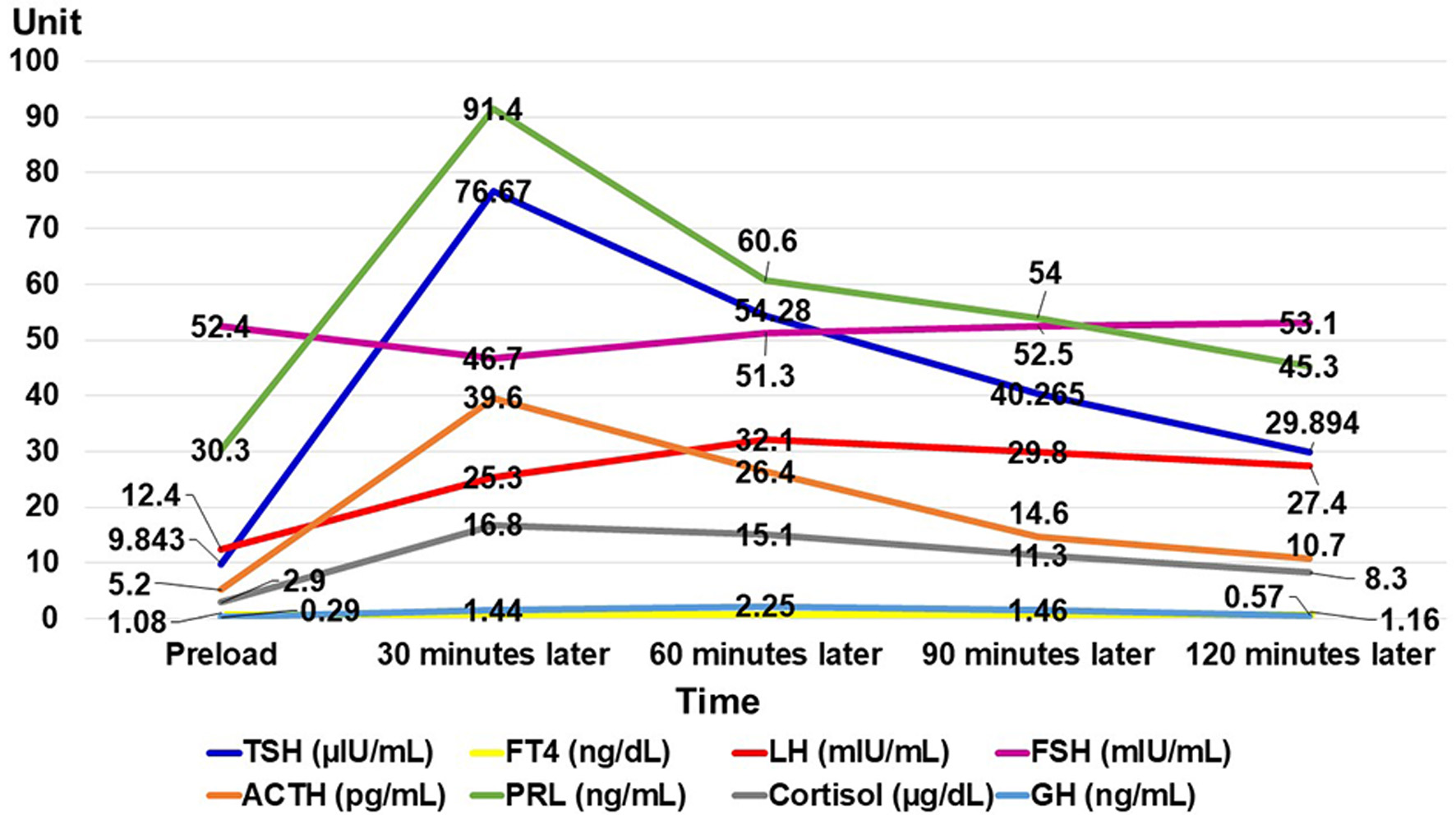 Click for large image | Figure 5. Result of anterior pituitary function test through combined intravenous administration of four hypothalamic releasing hormones. A FT4 test was performed twice: preload, and 120 min later. TSH: thyroid-stimulating hormone; FT4: free thyroxine; LH: luteinizing hormone; FSH: follicle-stimulating hormone; ACTH: adrenocorticotropic hormone; PRL: prolactin; GH: growth hormone. |
Based on these results, we suspected the following pathology: CSF filled her sella turcica and caused insufficient pituitary hormone secretion. Then, she suffered from adrenal gland cortex function failure, and diurnal variation of cortisol vanished based on low ACTH secretion. On the other hand, cortisol secretion had been sustained at the very limit, and a time lag occurred between cortisol and ACTH secretion. The phenomenon was due to PES. Along the way, some kinds of stress had caused further ACTH hyposecretion, which led to her symptoms. Therefore, she only ever experienced hyponatremia due to temporary adrenal insufficiency when she experienced stress. Her gonadal hormone function had been maintained because she hadn’t experienced amenorrhea, and she had given birth. The cause of her complete blindness was suspected to be due to compression of the optic nerve by ES. On the other hand, the cause of ES in this case remained unclear.
Although we could not get a definite diagnosis, we diagnosed it as adrenal insufficiency due to inappropriate ACTH secretion caused by PES. We concluded that it would be safer to treat her through corticosteroid replacement therapy, especially in the morning, because she would repeatedly suffer from adrenal insufficiency with high percentages under stress conditions. We started hydrocortisone (15 mg/day) from the 10th hospital day, and she immediately became symptom-free, and she was discharged on the 19th hospital day. On the other hand, we also expected to administer hydrocortisone (10 mg) to be taken in the event of stress, including similar symptoms, burn, or trauma.
| Discussion | ▴Top |
We present the first known case of adrenal insufficiency due to insufficient ACTH secretion caused by total PES.
ESS is considered to a relatively rare entity, but research reports that it is present in 5.5-20% of autopsies, and that it is also present in an estimated 12% of neuroimaging patients [3, 19]. Additionally, some reports note an even higher incidence in clinical practice, with estimates of up to 35% [3]. ESS incidence peaks in the forties through sixties [3, 9]. However, the incidence of pediatric ES varies significantly by population surveyed, ranging from as little as 1.2% in children without endocrine symptoms to as much as 68% in children with known endocrinopathy [2].
The prevalence of PES (i.e., ES with no discernible cause) is not precisely known; estimates range from 2-20% [1]. In a clinical analysis of 123 cases of PES in China, the average age of (43 males, 80 females) PES patients was 59.2 ± 13.6 years (ranging 24 - 92 years), among whom 61% of the patients were in the age group 50 - 69 years old [6]. It was demonstrated that there is a female predisposition with a ratio of 4:1 or 5:1 over males, in females with a history of multiple pregnancies, and that PES is more common in obese patients [3, 9].
Endocrine abnormalities are present in less than 20% of cases of ES [3]. Although pituitary hormonal dysfunction is more commonly seen in women, it appears to occur more frequently in men with ES [3]. 50% of patients present with multiple hormonal deficiencies; among these, the endocrine abnormality mostly commonly found in PES patients is hyperprolactinemia [6]. In addition, among patients with PES, it is estimated that 1 in 80,000 individuals has central hypothyroidism [6].
ES is usually discovered in the course of brain imaging studies performed for separate indications, and in most cases, the condition is asymptomatic [20]. Patients with compromised pituitary function may have histories and physical exams that are consistent with any or all pituitary hormone deficiencies [3]. ES may result in impairments to a variety of endocrine glands, for which the pituitary gland produces crinins [20]. Despite the relatively high incidence of ESS (as much as 5% of the population as a whole), it is commonly ignored as a cause of various symptoms [20]. ES complications may potentially include partial or complete hypopituitarism, or hormone hyperfunctioning, including hyperprolactinemia-induced infertility in ES [3, 21]. Though ES may be largely asymptomatic in adult patients, in children it is more likely to be associated with clinical symptoms and endocrinopathies, particularly GH deficiency, hypogonadotropism, or multiple pituitary hormone deficiencies [2, 22].
Because endocrine function is ordinarily intact, patients with ESS typically have normal histories and physical exams [3]. Clinical manifestations of ESS include headache (the most typical manifestation), hypopituitarism, CSF rhinorrhea, visual abnormalities and deterioration including impairment of the visual field, overweight/obesity, hypertension, irregular menses, primary amenorrhea, and multiple pregnancies, some of which may often be associated with intracranial hypertension [3, 5, 8, 9, 23-25]. In addition, there are reports of episodes of vertigo, dizziness, and hearing loss; osteoporotic fractures; persistent non-fusion of the hand epiphyses; and bradycardia [10]. One study found that frequent symptoms of ESS patients included fatigue (56.1%), headache (34.1%), nausea and vomiting (17.9%), gonadal dysfunction (17.1%), visual abnormalities (5.7%), and hypopituitarism crisis (3.3%) [26]. Additionally, another paper reported that hypopituitarism had been found in 54% of patients, and among these, 36.6%, 31.7% and 17.1% had been patients with central hypoadrenalism, hypogonadism, and hypothyroidism, respectively [26]. Although hyperprolactinemia and hypopituitarism, including GH or other hormone deficits, represent the most common endocrine abnormalities in both male and female patients, ESS is typically heterogeneous in both its clinical manifestation and hormonal alterations, and can sometime reach severe extremes, leading to cases of papilledema, CSF rhinorrhea, or reduced visual acuity [8, 9]. Naturally, the occurrence of hypopituitarism in patients with complete ESS was significantly higher than that in those with partial ESS (P < 0.05) [26]. Furthermore, in 13% of patients, ESS was concomitant with other autoimmune diseases, including 9% with Graves’ disease and 2% with Cushing’s syndrome due to adrenal adenoma [26]. On the other hand, there may be increased incidences of hypertension, CSF rhinorrhea, and pseudotumor cerebri among ESS patients [6]. Additionally, for ESS patients, hypopituitarism should be considered in all patients with syndrome of inappropriate secretion of antidiuretic hormone (SIADH) like the clinical picture with no apparent cause (e.g., bronchogenic carcinoma), and even in patients who demonstrate mild dehydration with inappropriately low serum sodium [6]. Severe hyponatremia should alert physicians to Addisonian crises [6]. Recurrent manic-like episodes can also be induced by hyponatremia, possibly as a result of ESS [27]. In our case, recurrent manic-like episodes had been confirmed, and hyponatremia was seen upon her admission; therefore, we suspected adrenal cortical insufficiency as the cause.
For a diagnosis of ESS, a careful history and physical examination should first be conducted, with regards to symptoms of hypopituitarism [6]. In addition, because ES is a radiologic finding and diagnosis, the differentials relating to this entity stemming from each specific cause (e.g., intracranial hypertension) should be considered [1, 3, 5]. It is necessary to consider the significance of using a combination of clinical findings, pituitary gland function examination, and brain imaging study when scanning to check for a diagnosis of ESS [5, 7, 17, 28]. In most patients, ESS was initially discovered as an incidental finding through imaging studies, and in nearly a quarter of patients, ESS was discovered during diagnostic anterior pituitary deficiency evaluation, which is more common in men [9]. On the other hand, children with ES findings require endocrinologic and ophthalmologic evaluation [2].
There have been significant advances in pituitary imaging over the past few decades: most diagnoses are made through an MRI scan, and pituitary imaging is recommended in patients who have pituitary hormone excess, hypopituitarism, or mass effect in the sella [9, 29]. Modern imaging modalities have vastly improved our ability to detect and characterize sellar masses, and to characterize the extent and spread of lesions in and around the sella more accurately [29]. In addition, intraoperative MRI scans may help increase sellar mass resection completeness [29]. Other imaging modalities, such as magnetic resonance angiography, CT scans, and CT angiography, also play important roles in specific cases [29].
An overall pituitary function evaluation was essential for patients with headache and fatigue, or with suspected ESS [26]. In addition, although hormone deficiencies are found in only a small fraction of ESS patients, a full pituitary hormone workup is the indicated screening approach at diagnosis, and patients should be referred to an endocrinologist [3]. Therefore, it is advisable to be aware of the need for hormone level evaluations at the time of diagnosis, and for diagnosis purposes, testing of the entire pituitary axis is appropriate and highly recommended in patients with ESS [1, 3]. However, there are currently no society-based guidelines, and it remains unclear whether, and to what extent, asymptomatic adult patients in which ES was incidentally discovered should undergo hormonal disturbance diagnostic testing [1]. In most cases, pituitary function is normal despite the pituitary gland’s abnormal appearance of the pituitary gland, but in approximately 20% of cases, any or all pituitary hormone levels may be affected [3]. Hyperprolactinemia and GH deficiency appear to be the two most common findings in ES: hyperprolactinemia is present in 10-17% of cases and may be due to a microprolactinoma or functional hyperprolactinemia, while GH deficiency is present in 4-60% of cases, but its clinical significance in adults is unclear [3]. Gonadotropin deficiency is found in 2-32% of cases, while ACTH, thyroid-stimulating hormone (TSH), and antidiuretic hormone (ADH) deficiencies are less frequent, with incidences of approximately 1% each [3]. Specifically, in some reports, authors advised following basic neuroendocrinological testing: fasting cortisol, free thyroxine (FT4), estradiol or testosterone, insulin-like growth factor 1 (IGF-1), and prolactin [1]. In addition, the following labs are necessary for pituitary function evaluation in any patient found to have ES: for the adrenal axis, early-morning fasting cortisol levels are a screening option for ACTH deficiency, and overtly low levels of cortisol (less than 3.0 µg/dL) are considered consistent with adrenal insufficiency [3]. Morning cortisol levels greater than 11.0 (or, some authors suggest, 14.0) µg/dL suggest that adrenal insufficiency is highly unlikely, while morning cortisol levels of 3.1 to 11.0 (14.0) µg/dL are indeterminant and can warrant further testing, such as ACTH stimulation testing [3]. If morning cortisol levels are low, an ACTH level should be obtained and correlated to the low morning cortisol levels, to help differentiate primary from secondary/central adrenal insufficiency [3]. Metyrapone testing can also be of assistance in this diagnosis [3]. If corticosteroid excess is suspected, an ACTH level needs to be once more correlated with cortisol levels, and a workup for Cushing’s syndrome should be the next step [3]. From this point of view, in this case, morning cortisol levels were 7.3 µg/dL: indeterminant adrenal insufficiency is suspected, and further testing such as ACTH stimulation testing is warranted.
If pituitary tropic hormones are inappropriately low in the presence of low target hormones, physicians should screen for secondary endocrine insufficiencies [6]. Moreover, it is easy to overlook pituitary deficiencies in the event that only tropic hormones are used for hormone deficiency evaluations [6]. In particular, it is difficult to diagnose hyponatremia due to isolated ACTH deficiency, as it is ordinarily indistinguishable from non-endocrine SIADH [30]. For microadenoma, visual field testing and screening for hypopituitarism are not considered necessary, but in the event that it is associated with ES, both visual field testing and screening for hypopituitarism are necessary [19]. In this case, we ultimately diagnosed it as total PES, because total pituitary gland thickness was less than 2 mm, and over 50% of the sella was filled with CSF; however, the etiology was unclear.
In most cases, no treatment is necessary for ESS [3]. ESS treatment includes replacement of hormone deficiencies, with occasional surgical measures for the relief of obstructive intracranial lesions [2]. A multidisciplinary approach, integrating endocrine, neurologic and ophthalmologic expertise, is strongly advocated, and is recommended for the proper diagnosis, management, treatment, and follow-up of ESS and all related abnormalities [8].
Most patients with ES remain asymptomatic throughout their life and require no treatment; however, in cases where isolated ACTH deficiency develops, corticosteroid treatment should be enforced in order to avoid fatal consequences [30]. Patients allergic to succinate ester can tolerate alternative ester-free corticosteroids [31]. Patients with hypopituitarism should be given hormone replacement therapy in time, and followed up afterwards, but chronic hyponatremia should always be corrected gradually, in order to avoid osmotic myelinolysis syndromes [26, 32].
When patients with ES findings present with visual impairment or deterioration, a surgical treatment may be necessary [22]. The main goal of this surgery is to elevate sellar content through a transsphenoidal approach [22]. Currently, chiasmapexy is an effective surgical method for treating visual deterioration caused by ESS, to correct the downward displacement of the suprasellar visual system into an ES, which causes visual impairment, and various materials are used for the elevation of the optic chiasm [33-35]. Intradural chiasmapexy is indicated in treatment of SES, while the extradural approaches are suggested for surgical management of PES [33]. Endoscopic endonasal chiasmapexy with septal cartilage and sphenoidal sinus bone is also worth considering as an option, as it is minimally invasive, and involves a decreased risk of infection [34]. However, the use of artificial substances may bring a risk of graft infection, and after surgery, fat and muscle may be absorbed over the long term [35]. There is also the possibility that bone and cartilage may be unavailable in sufficient quantities [35]. One advantage of iliac bone is its reduced likelihood of absorbing and becoming infected compared to synthetic materials [35]. This approach may be suitable for reoperative cases, especially in patients whose septal cartilage has been removed in a previous surgery [35]. This method halts visual deterioration and may be worth considering as an option for chiasmapexy operations [35]. On the other hand, another report introduced a novel technique for the precise reconstruction of the sellar floor, using a heterologous bone block to restore the anatomic elements of the sella turcica; the technique was simple and reproducible, and allowed for nearly exact and persistent elevation of sellar content [22]. Furthermore, intraoperative MRI scans help to achieve adequate sellar packing while avoiding both insufficient packing and overpacking [36]. According to some reports, bariatric surgery may be effective in intracranial hypertension treatment; however, no data exists regarding ESS [18].
As a prognosis, the presence of ESS does not affect life expectancy, as it is generally a benign condition [3]. On the other hand, when a specific hormone deficiency or excess is present, the prognosis varies depending on the specific hormone abnormality and its treatment [3]. In addition, because ES or pituitary atrophy and endocrine impairments can manifest even decades after radiation and chemotherapy, patients of these should undergo regular follow-ups, including pituitary MRI scans and hormonal examinations [37]. Ultimately, the potentially lethal outcome of pituitary infection makes the correct diagnosis and therapy critical [16].
This case study has several limitations. First, this paper reviews a single case report and case series of ESS. Therefore, the actual situation of ESS may differ from the results of the literature review, due to reporting bias. Second, we did not measure hormone values during the patient’s convalescent stage; therefore, we should have measured them and compared them with the values at time of admission for a detailed diagnosis. Third, metyrapone testing should be performed to assist in this diagnosis.
In conclusion, we have reported the first known case of adrenal insufficiency due to insufficient ACTH secretion caused by total PES. This case highlights the complex pathology, procedure of diagnosis, and treatment for ESS. On the other hand, further investigations are needed to clarify the precise pathogenesis of ESS.
Acknowledgments
The author would like to thank Dr. Kento Takeshima, from the Department of General Medicine, Kanazawa Medical University Himi Municipal Hospital, for her invaluable help with review of the literature.
Financial Disclosure
None to declare.
Conflict of Interest
The authors declare that they have no competing interest.
Informed Consent
Both written and verbal informed consents were obtained from the patient for publication of this case report and any accompanying images.
Author Contributions
D Usuda collected the case data, prepared the photos, and wrote the manuscript. All authors proofread the pathologic materials. S Takagi, K Takanaga, T Izumida, R Sangen, T Higashikawa, and Y Kasamaki proofread and revised the manuscript. All authors approved the final version to be published.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
ES: empty sella; CSF: cerebrospinal fluid; ESS: ES syndrome; PES: primary ESS; SES: secondary ESS; ACTH: adrenocorticotropic hormone; CT: computed tomography; MRI: magnetic resonance imaging; ITT: insulin tolerance test; CRH: corticotropin-releasing hormone; TRH: thyrotropin-releasing hormone; LH-RH: luteinizing hormone-releasing hormone; GRF: growth-hormone releasing factor; GH: growth hormone; SIADH: syndrome of inappropriate secretion of antidiuretic hormone; TSH: thyroid-stimulating hormone; ADH: antidiuretic hormone; FT4: free thyroxine; IGF-1: insulin-like growth factor 1
| References | ▴Top |
- Auer MK, Stieg MR, Crispin A, Sievers C, Stalla GK, Kopczak A. Primary empty sella syndrome and the prevalence of hormonal dysregulation. Dtsch Arztebl Int. 2018;115(7):99-105.
doi pubmed - Lenz AM, Root AW. Empty sella syndrome. Pediatr Endocrinol Rev. 2012;9(4):710-715.
- Ucciferro P, Anastasopoulou C. Empty Sella. In: StatPearls. Treasure Island (FL), 2020.
- Izizag BB, Ngandu A, Mbiso DL. [Empty sella syndrome: a case report]. Pan Afr Med J. 2019;33:317.
doi pubmed - Braiteh N, Breiteh N, Kashou HE. Sinus bradycardia as a rare and unusual presentation of partial empty sella syndrome: a case report. Am J Case Rep. 2019;20:1071-1074.
doi pubmed - Aijazi I, Abdullah Al Shama FM, Adam Mukhtar SH. Primary empty sella syndrome presenting with severe hyponatremia and minimal salt wasting. J Ayub Med Coll Abbottabad. 2016;28(3):605-608.
- Jurca MC, Bembea M, Kozma K, Sandor MI, Negrean RA, Dobjanschi L, Cuc EA, et al. Empty sella associated with growth hormone deficiency and polydactyly. Rom J Morphol Embryol. 2018;59(1):381-384.
- Chiloiro S, Giampietro A, Bianchi A, Tartaglione T, Capobianco A, Anile C, De Marinis L. DIAGNOSIS OF ENDOCRINE DISEASE: Primary empty sella: a comprehensive review. Eur J Endocrinol. 2017;177(6):R275-R285.
doi pubmed - Guitelman M, Garcia Basavilbaso N, Vitale M, Chervin A, Katz D, Miragaya K, Herrera J, et al. Primary empty sella (PES): a review of 175 cases. Pituitary. 2013;16(2):270-274.
doi pubmed - Delgado-Hernandez A, Verduzco-Mendoza A, Luna-Reyes FA, Marquez-Palacios S, Arch-Tirado E. [Analysis of the joint and a posteriori probability between primary empty sella, its comorbidities and audiovestibular pathology]. Cir Cir. 2015;83(6):459-466.
doi pubmed - Gonzalez-Tortosa J, Piqueras-Perez C, Ruiz-Espejo A, Martinez-Lage JF. [Reversible primary empty sella. Case report]. Neurocirugia (Astur). 2010;21(4):317-321.
doi - Colao A, Cotta OR, Ferone D, Torre ML, Ferrau F, Di Somma C, Boschetti M, et al. Role of pituitary dysfunction on cardiovascular risk in primary empty sella patients. Clin Endocrinol (Oxf). 2013;79(2):211-216.
doi pubmed - Chang J, Tran J, Kamel D, Basu A. Nivolumab-induced hypophysitis leading to hypopituitarism and secondary empty sella syndrome in a patient with non-small cell lung cancer. BMJ Case Rep. 2019;12(3):e228135.
doi pubmed - Gao H, Gu YY, Qiu MC. Autoimmune hypophysitis may eventually become empty sella. Neuro Endocrinol Lett. 2013;34(2):102-106.
- Kusano Y, Horiuchi T, Oya F, Miyaoka Y, Oguchi K, Takemae T, Hongo K. Ectopic pituitary adenoma associated with an empty sella: a case report and review of the literature. J Neuroimaging. 2013;23(1):135-136.
doi pubmed - Pepene CE, Ilie I, Mihu D, Stan H, Albu S, Duncea I. Primary pituitary abscess followed by empty sella syndrome in an adolescent girl. Pituitary. 2010;13(4):385-389.
doi pubmed - Chen H, Li Y, Zhang P, Wang Y. A case report of empty Sella syndrome secondary to Hantaan virus infection and review of the literature. Medicine (Baltimore). 2020;99(14):e19734.
doi pubmed - D'Alessandris QG, Montano N, Bianchi F, Doglietto F, Fernandez E, Pallini R, Lauretti L. Persistence of primary empty sella syndrome despite obesity surgery: report of two unusual cases. Br J Neurosurg. 2012;26(6):875-876.
doi pubmed - Agrawal NK, Jain P, Garg S. Primary empty sella with isolated ACTH deficiency and microprolactinoma. Gynecol Endocrinol. 2012;28(7):568-569.
doi pubmed - Holecki M, Rembiesa-Jarosinska E, Fryzlewicz-Moska A, Dulawa J. Diagnostic dilemmas in a patient with anaemia. Empty sella syndrome—a case report. Endokrynol Pol. 2010;61(4):400-403.
- Kazama T, Yasuda K, Kimura H. [Infertility due to hyperprolactinemia in empty sella : a case report]. Hinyokika Kiyo. 2019;65(2):55-59.
- Guinto G, Nettel B, Hernandez E, Gallardo D, Arechiga N, Mercado M. Osseous remodeling technique of the sella turcica: a new surgical option for primary empty sella syndrome. World Neurosurg. 2019;126:e953-e958.
doi pubmed - Sivaraju L, Thakar S, Hegde AS. Visual Deterioration and Herniation of the Anterior Cerebral Artery: Unusual Presentation of an Empty Sella Syndrome Complicating Decompression of a Rathke Cleft Cyst. J Neuroophthalmol. 2016;36(2):156-158.
doi pubmed - Xu P, He H, Chen Y, Wang C, Zhu Y, Ye X. Osteoporotic fractures and persistent non-fusion of the hand epiphyses caused by empty sella syndrome in an adult: a case report. J Int Med Res. 2013;41(5):1768-1772.
doi pubmed - Dange N, Redhu R, Kawale J, Mahore A. Primary amenorrhea due to empty sella: an underestimated entity. Turk Neurosurg. 2012;22(4):499-501.
doi - Li J, Jia HW, Wang CL, Zhang R, Qu MY, Li W, Yuan MH, et al. [A clinical analysis of 123 cases of primary empty sella]. Zhonghua Nei Ke Za Zhi. 2017;56(4):268-272.
- Yang CH, Lin YC, Chou PH, Chen HC, Chan CH. A case report of late onset mania caused by hyponatremia in a patient with empty sella syndrome. Medicine (Baltimore). 2016;95(6):e2629.
doi pubmed - Saindane AM, Lim PP, Aiken A, Chen Z, Hudgins PA. Factors determining the clinical significance of an "empty" sella turcica. AJR Am J Roentgenol. 2013;200(5):1125-1131.
doi pubmed - Faje A, Tritos NA, Swearingen B, Klibanski A. Neuroendocrine disorders: pituitary imaging. Handb Clin Neurol. 2016;136:873-885.
doi pubmed - Doroftei NA, de Rudder C, de Visscher N, Hanon F. Isolated ACTH deficiency in a patient with empty sella as revealed by severe hyponatremia. Acta Clin Belg. 2016;71(6):451-454.
doi pubmed - Nucera E, Lombardo C, Aruanno A, Colagiovanni A, Buonomo A, de Pasquale T, Pecora V, et al. 'Empty sella syndrome': a case of a patient with sodium succinate hydrocortisone allergy. Eur J Endocrinol. 2011;164(1):139-140.
doi pubmed - Gupta VA, Karnik N, Itolikar M, Vekariya K. Extra-pontine myelinolysis in a case of pan-hypopituitarism due to empty sella syndrome. J Assoc Physicians India. 2015;63(10):86-87.
- Barzaghi LR, Donofrio CA, Panni P, Losa M, Mortini P. Treatment of empty sella associated with visual impairment: a systematic review of chiasmapexy techniques. Pituitary. 2018;21(1):98-106.
doi pubmed - Ishihara E, Toda M, Sasao R, Ozawa H, Saito S, Ogawa K, Yoshida K. Endonasal chiasmapexy using autologous cartilage/bone for empty sella syndrome after cabergoline therapy for prolactinoma. World Neurosurg. 2019;121:145-148.
doi pubmed - Tsukiyama A, Hattori Y, Tahara S, Ishisaka E, Morimoto D, Oyama K, Teramoto A, et al. New technique for chiasmapexy using iliac crest bone graft: 2 cases of visual impairment caused by empty sella syndrome. World Neurosurg. 2017;107:1051 e1019-1051 e1025.
doi pubmed - Kubben PL, Cornips EM, Looij BJ, Beuls EA. Transsphenoidal treatment of secondary empty sella syndrome using low field strength intraoperative MRI: case report. Minim Invasive Neurosurg. 2010;53(4):198-202.
doi pubmed - Nishi Y, Hamamoto K, Fujita N, Okada S. Empty sella/pituitary atrophy and endocrine impairments as a consequence of radiation and chemotherapy in long-term survivors of childhood leukemia. Int J Hematol. 2011;94(4):399-402.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.