

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website http://www.jofem.org |
Original Article
Volume 1, Number 4, October 2011, pages 174-181
Compound Heterozygous Mutations in Vitamin D Receptor Gene in Two Sisters With Hereditary Vitamin D Resistant Rickets Type II
Samar El Sodaa, b, d, Hanan Madanic, e
aPediatric Medicine, Cairo University, Egypt
bDepartment of Pediatrics, Fakhry and Al Rajhi Hospital, Al Khobar, Kingdom of Saudi Arabia
cMolecular Unit of the Chemical Pathology Laboratories, Department of Clinical Pathology, Faculty of Medicine, Cairo University, Kasr El Aini Street, Cairo, Egypt
dCorresponding author: Department of Pediatrics, Fakhry and Al Rajhi Hospital, Al Khobar, Kingdom of Saudi Arabia, 31952, P.O. Box 251
eCorresponding author: Department of Chemical Pathology, Faculty of Medicine, Cairo University, Kasr El Aini Street, Cairo, Egypt
Manuscript accepted for publication October 12, 2011
Short title: Mutations in Gene
doi: https://doi.org/10.4021/jem41w
| Abstract | ▴Top |
Background: Hereditary vitamin D resistant rickets (HVDRR) type II, is a rare autosomal recessive disorder with known heterogeneity in clinical features, response to treatment and presence or absence of vitamin D receptor (VDR) gene mutation. Here, we described VDR gene mutations in 2 sisters with HVDRR with correlation to clinical features, and response to treatment.
Methods: Vitamin D receptor gene sequence analysis was performed for exons 2 and 3, in addition to polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) for exons 7 and 8. Patients, their parents, their elder sister and brother were studied. Treatment with high dose vitamin D, response to therapy, and 30 months follow up were recorded for both patients.
Results: A novel heterozygous point mutation (c.A155G) at the initiation codon of exon 3 was found in the 2 patients, their father and their healthy brother. This point mutation changed the amino acid (aa) at position 51 of the VDR from serine to glycine (p.Ser51Gly) at the start of the second zinc finger of the DNA-binding domain of the VDR. Exon 7 showed a heterozygous mutation in both patients and their mother. The mutation was located at the 970 base pair (bp) of VDR. Patients responded well to high doses oral calcium and Alfacalcidol without relapse of their rickets for the whole follow up period.
Conclusion: Compound heterozygous mutation was identified in the VDR gene in 2 sisters with HVDRR. This mutation resulted in HVDRR without alopecia that was responsive to high dose vitamin D therapy.
Keywords: HVDRR; VDR gene mutation; Alopecia; Compound heterozygous mutation
| Introduction | ▴Top |
Hereditary vitamin D resistant rickets (HVDRR) type II, is a rare autosomal recessive disorder most often caused by mutations in vitamin D receptor (VDR) gene, leading to end organ resistance to 1,25 dihydroxyvitamin D (1,25(OH)2D3) and resulting in increased circulating level of 1,25(OH)2D3 [1]. Patients present with early onset rickets, hypocalcemia, hypophosphatemia, increased serum calcitriol levels and secondary hyperparathyroidism and some patients develop total body alopecia [2]. Data from several studies have shown heterogeneity in HVDRR regarding clinical features, cellular defects in cultured skin fibroblasts [3], presence and type of VDR gene mutation [4]. This diversity has been evidenced clinically by the various responses to treatment with high doses of vitamin D analogues and by the presence or absence of alopecia [5].
VDR is a member of the steroid, thyroid, retinoid receptor gene superfamily of nuclear transcription factors. VDR is composed of a N-terminal DNA-binding domain (DBD) that enables the interaction with vitamin D response elements (VDREs) and a C- terminal ligand-binding domain (LBD) that binds to 1,25(OH)2D3 [1, 6]. VDR gene is located on chromosome 12q13.11 and spans approximately 75 kb of genomic DNA [7]. About 40 different mutations in the VDR gene have been described to date in approximately seventy families with HVDRR-type II [8].The majority of these mutations were point mutations, while others included deletion, insertion, and duplication mutations involving multiple base pairs.
The aim of our study was to identify the VDR gene mutation in two siblings with HVDRR and to correlate the type and site of this mutation with the phenotypic expression, clinical presentation, and response to treatment in the 2 patients. We expected such work to help derive further information on vitamin D resistance.
| Subjects and Methods | ▴Top |
Description of Subjects
We studied a family consisting of 6 subjects: the father, the mother, 2 healthy siblings and 2 affected patients. They are of Middle-eastern origin. The 2 patients are the third and fourth issue of a non consanguineous marriage between a Saudi father and a Bahraini Mother. Both parents are phenotypically normal. They have an apparently normal elder sister and brother, 17 and 14 years old respectively. Both patients were the outcome of uneventful pregnancy and delivery. They were healthy at birth and had normal development for the first few months of life. In the second half of their first year they were noted to be small for age and slow in developing motor skills. None of the affected children had alopecia. They received physiological doses of different forms of vitamin D and calcium supplementation without improvement. The elder sister underwent surgical correction of bone deformity twice, but was followed by recurrence of bowing. Patients were first seen in January 2008. Laboratory (Table 1) test was done as well as x-rays long bones (Fig. 1). Due to social reasons patients did not come back for follow up until February 2009. Both of them were off treatment for the previous 13 months. Laboratory (Table 1) and radiological (Fig. 1) work up was repeated.
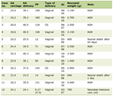 Click to view | Table 1. Biochemical and hormonal profiles of the 2 patients |
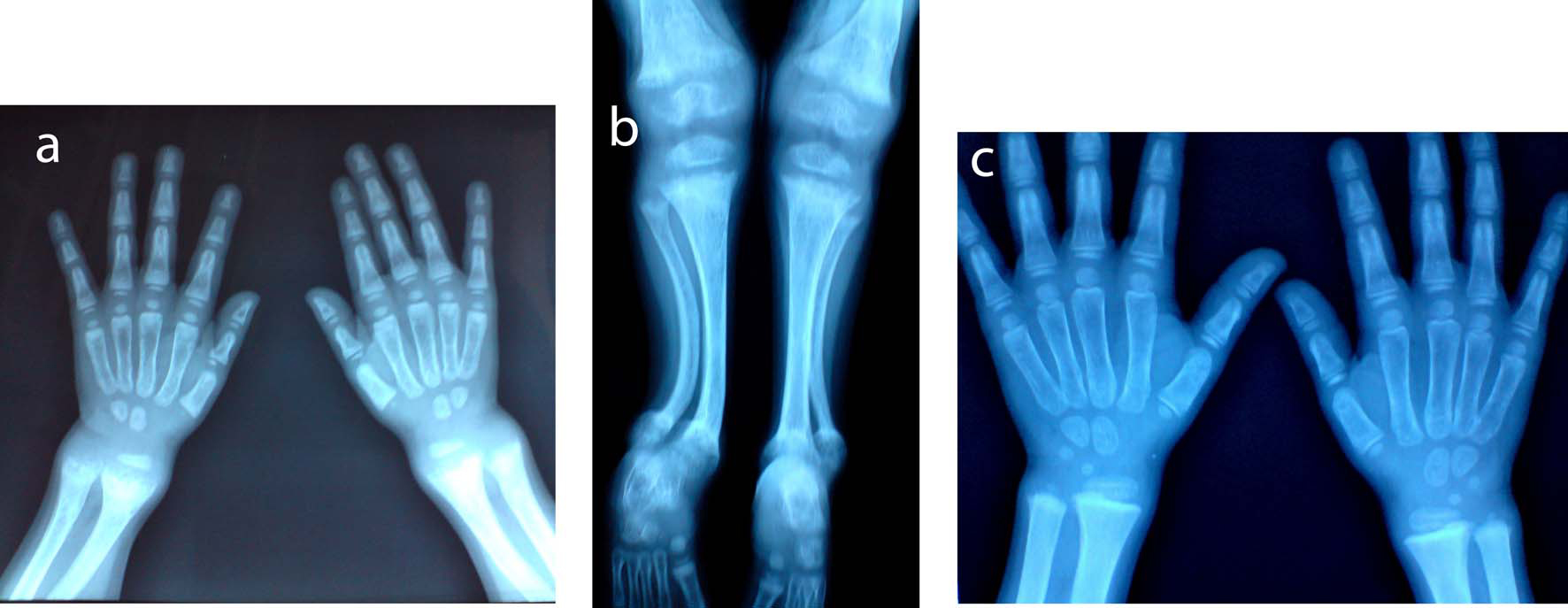 Click for large image | Figure 1. X-ray A) wrist and B) lower limb of the first patient showed radiological signs of active rickets: diffuse osteoporosis, widening of the zone of provisional calcification at the metaphysis, metaphyseal fraying, flaying and cupping of proximal and distal tibia, distal femur, radius and ulna, irregular ill defined outline of the epiphysis, genu valgum deformity, bilateral anterior bowing of the lower part of the tibia and fibula, and delayed bone age. C) Improvement of signs of active rickets and of bone age after treatment. |
The first patient is a nine–year-old female, her height was 111 cm, below the 5th percentile for age, weight 25.5 kg on the 25th percentile for age. She had broadening of the lower end of radius and ulna, chest deformity, marked bowing of the lower limb (genu varum), waddling gait, normal muscle tone and normal dentition. She was complaining of marked lower limb pain that prevented her from walking, playing or joining sport class. Her x-ray showed diffuse osteopenia, widening of the epiphyseal plates, enlarged distal femoral epiphyses, bilateral genu varum deformity, but no flaying, cupping, or fraying of any of the metaphyseal ends of long bones. One year later her height was 114 cm (still below the 5th percentile for age), and her weight was 30 kg (above the 25th percentile for age). Her x-ray showed only genu varum deformity and improvement of previously noted osteopenia. Mean bone densitometry measured at lumbar vertebra (L1, L2, L3, L4) showed normal bone density: 201.4 mg/cm2 (normal > 150 mg/cm2).
The second patient is a three-year-old girl with height and weight below the 5th percentile for age (73cm and 7.8 kg respectively). She had features of florid rickets manifested as broadening of the lower ends of long bones at the wrist and ankle, pigeon chest, rachitic rosary, genu valgum (knock knee), bilateral tibial kyphosis, normal dentition for age, and normal muscle tone. The marked lower limb deformity had markedly limited her motor activity. One year later her height and weight were 75 cm and 9.5 kg respectively (below the 5th percentile for age). She was still having the same clinical and radiological signs of active rickets. She had premature loss of the lower central incisors. Mean bone densitometry measured by CT at lumbar vertebra (L1, L2, L3, L4) showed marked osteoporosis: 101.4 mg/cm2 (normal >150 mg/cm2) (Fig. 1A, 1B, 1C).
| Methods | ▴Top |
Laboratory and radiological work up was consistent with the diagnosis of HVDRR type II. Treatment started in February 2009 with oral Alfacalcidol (one alpha drops) (2 mcg/day) that was gradually increased in increments of 0.5 mcg every 3 - 4 weeks in addition to oral calcium supplementation in the form of 1700 mg calcium glubionate daily (this was present in a combination with 800 IU of active form of vitamin D3). Follow up was done every 2 weeks for the first 3 months and monthly thereafter until normalization of biochemical results and x-ray.
Alfacalcidol was then reduced gradually and later discontinued, while oral calcium supplementation continued throughout the study period. Follow up continued every 2 - 3 months up to a total of 30 months. Signs of vitamin D toxicity were monitored by 24 hours urinary calcium excretion, calcium / creatinine ratio, serum creatinine, blood urea nitrogen and renal ultrasonography. Biochemical tests were repeated at every follow up visit. X-ray and bone densitometry were repeated after normalization of biochemical tests. Height growth velocity was monitored throughout the study.
VDR gene analysis
Informed consent was obtained from parents for DNA study, but they refused skin biopsy of affected children. All family members underwent VDR DNA analysis. EDTA blood samples were used for VDR gene analysis using DNA sequencing and restriction fragment length polymorphism (RFLP). All molecular analyses were carried out at the Molecular Unit of the Chemical Pathology Laboratories, Faculty of Medicine, Cairo University, Egypt. Genomic DNA was extracted from peripheral blood using QiA amp DNA blood mini kit (Qiagen, Valencia, CA). Exons 2, 3, 7 and 8 of the VDR gene were amplified by polymerase chain reaction using the following primers and PCR conditions (Table 2). Exons 7 and 8 were amplified together & a pair of primers was designed to anneal to the intron-flanking regions of exons 7 and 8 at a site located 10-20 base-pair (bp) from the intron-exon boundaries [9].
Click to view | Table 2. Primers and PCR conditions used for amplifications |
Sequence analysis of exons 2 and 3
The amplified products for exons 2 and 3 (265, 220 bp respectively) were purified and were directly sequenced using the BigDye® Terminator v3.1 Cycle Sequencing Kit, and capillary electrophoresis was done on the Applied Biosystems 310. The nucleotide sequence obtained from the patients and their family was compared with hVDR complete cDNA sequence (Gene Bank accession NM_00107535.1) using the Blastn algorithm (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Data was then analyzed using CLC sequence viewer 6 program.
RFLP analysis of exons 7 and 8
The amplified products for exons 7 and 8 (573 bp) were analyzed by RFLP analysis, where the products were digested using RsaI restriction enzyme. Digestion of the normal PCR products with RsaI yielded 4 DNA fragments at 201, 195, 101 and 76 bp [1].
| Results | ▴Top |
Laboratory evidence of response to treatment was evident after one month of start of treatment by mild reduction of serum alkaline phosphatase (ALP). The first patient showed complete normalization of her biochemical tests (Table 1) with the exception of hypophosphatemia and normalization of PTH level after 11months of treatment at a dose of 7.5 mcg/day of Alfacalcidol. She continued to show hypophosphatemia throughout the treatment period and thereafter.
Gradual withdrawal of Alfacalcidol to 0.75 mcg/day was well tolerated but hyperparathyroidism and elevation of alkaline phosphatase recurred with discontinuation of one alpha. So patient was maintained on 0.75 mcg/day of one alpha for the remaining study period. Surgical correction was followed by normal bone healing. Her height and weight reached 117 cm and 36.5 kg respectively at the end of the first year of treatment, and 121 cm and 38 kg respectively by the end of the second year. No change was noted in growth velocity before and after treatment. Normal puberty occurred at the age of 11 years old.
The second patient showed complete normalization of all biochemical tests (Table 1) and PTH level at a dose of 4.5 mcg/day of Alfacalcidol after 5 months of regular treatment. Her x-ray showed healing of all signs of active rickets and improvement of osteopenia with persistence of the previously noted deformity. Bone mass density normalized to 184.7 mg/cm2 (normal above 150 mg/cm2). Dose of Alfacalcidol was then tapered to 0.7 mcg without recurrence of rickets. Patient then underwent surgical correction of lower limb deformity. She received the same dose throughout the post surgical period with normal bone healing and stable biochemical tests. Alfacalcidol was then discontinued and patient was maintained on oral 1700 mg of calcium glubionate and 800 IU of vitamin D3 daily for the last one year without recurrence of clinical or biochemical rickets. A marked acceleration of growth was noted during the treatment period. One year after starting treatment height reached 86 cm with an increase in growth velocity from 2 cm/year before treatment to 11 cm/year after starting treatment. Weight also increased to 12.5 kg and reached the 5th percentile for age. Height reached 96 cm at the end of follow up period. Patient had marked improvement in motor activity.
No signs of vitamin D toxicity were noted in the first patients, while the second patients showed polyuria with day and night incontinence that occurred when biochemical tests normalized. This was not associated with any change in the serum creatinine or blood urea nitrogen levels and was not associated with increase in urinary calcium / creatinine ratio. Polyuria improved with reduction of the dose of Alfacalcidol to 0.7 mcg/day.
Both parents had elevated PTH levels (120 and 88 pg/ml respectively), while other biochemical parameters were normal except for slight hypophosphatemia in the mother: PO4: 2.4 mg/dl (normal: 2.5 - 4.5 mg/dl). The healthy brother and sister had normal biochemical results.
Molecular genetic results
Direct sequencing of exon 2 showed a heterozygous mutation in exon 2 in the first patient only, where there is substitution of adenine by cytosine (A to C) at nucleotide position 22 of the VDR gene (Fig. 2). This point mutation leads to an altered amino acid at position 8, from threonine to praline (Fig. 2).
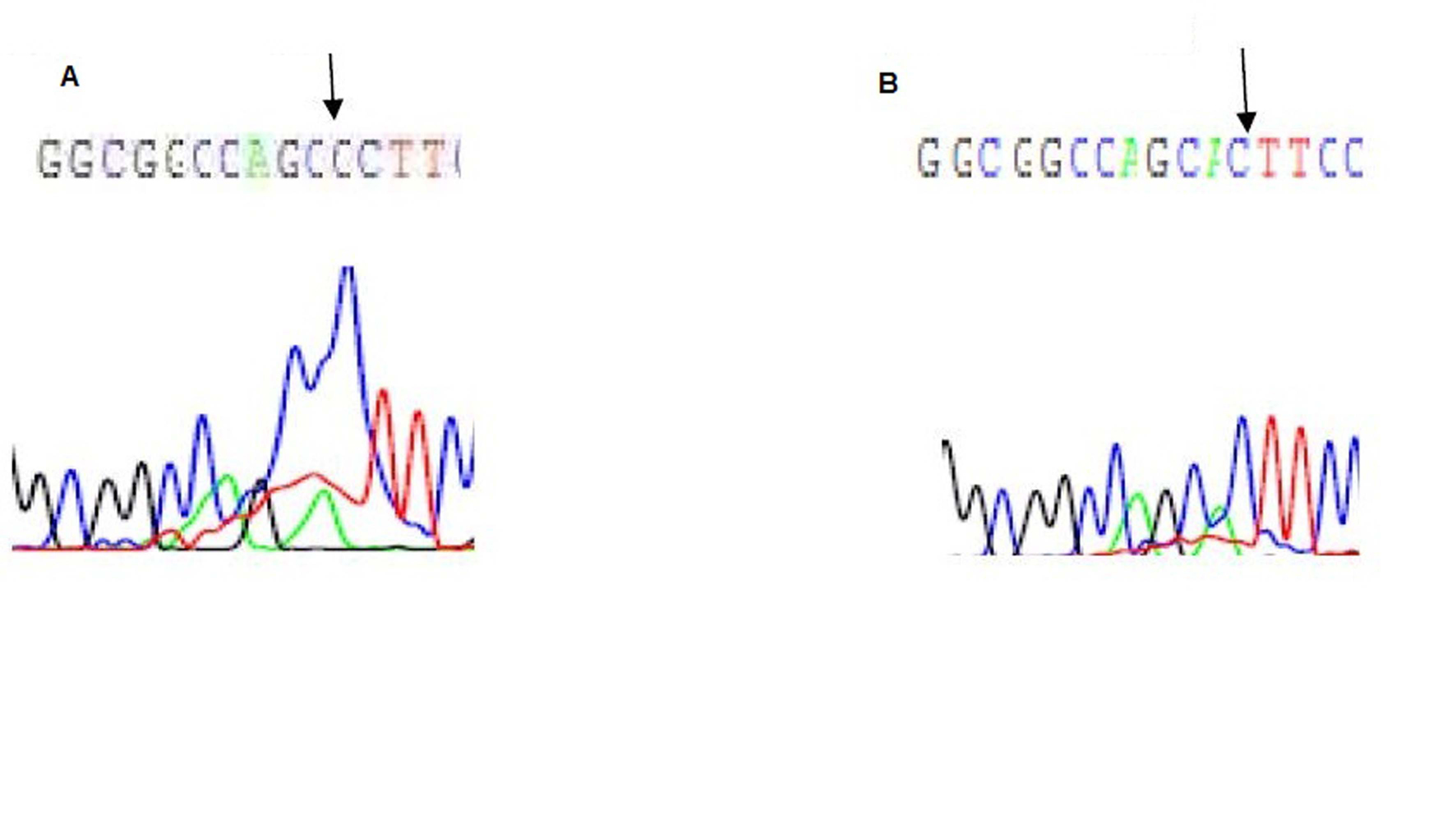 Click for large image | Figure 2. Electropherogram of exon 2 of the VDR gene: A is the first patient showing A to C substitution at position 22, B is the father with normal base sequence. |
Direct sequencing of exon 3 showed a missense mutation leading to heterozygous substitution of Adenine to Guanine (A-G) at the initiation codon of exon 3 at nucleotide position 155 of the VDR in patients, their father, and their phenotypically healthy brother. This results in changing the amino acid at position 51 of the VDR gene, from serine to glycine (Fig. 3).
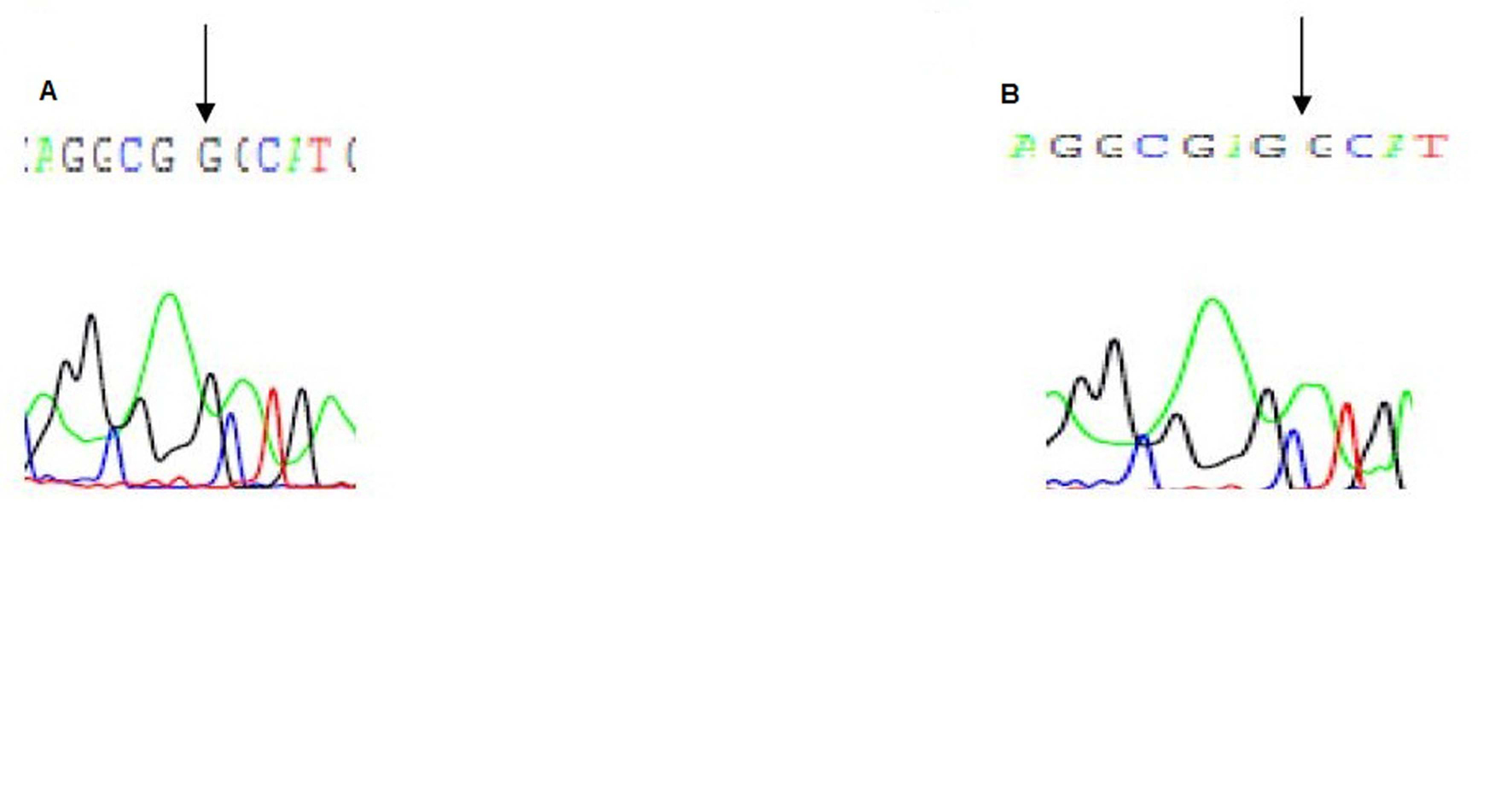 Click for large image | Figure 3. Electropherogram of exon 3 of the VDR gene: A is the first patient with a substitution and a deletion mutations, at nucleotide position 150 of the VDR gene, B is the father with the substitution mutation. |
PCR-RFLP analysis of exons 7 and 8 revealed heterozygous mutation in the 2 patients and their mother. The enzyme RsaI cuts at the GTAC sequence, which is altered by the mutation. The wild type allele shows a pattern of four bands 201, 195, 101, and 76 bp. This indicates that there are three RsaI sites in the normal DNA fragment. The mutation deletes one of the threeRsaI sites in the DNA fragment so that patients homozygous for the mutant allele exhibit only three 3 bands, 302, 195, and 76 bp. Heterozygote patients have both normal and mutant alleles i.e. 5 bands: 302, 201, 195, 101, and 76 bp.
| Discussion | ▴Top |
Patients described here exhibited the classical clinical pattern of HVDRR without alopecia [1, 2].
They initially presented with early onset rickets non responsive to physiologic doses of vitamin D, secondary hyperparathyroidism, and elevated serum 1,25(OH)2D3 levels in the first patient and high normal level in the second one. Although total serum calcium level was normal at presentation, we can suspect the presence of low ionized calcium that triggered the secondary hyperparathyroidism. Unfortunately test for ionized calcium was not available in our facility. The absence of very high level of 1,25(OH)2D3 initially in the second patient can be explained by the exhaustion of the 1 alpha hydroxylase system. This value increased after starting therapy when the patient changed to a new equilibrium.
Responses to pharmacological doses of vitamin D analogues have been reported in some patients [10-12]. However it has been shown that other patients may remain unresponsive to similar therapeutic trials [3, 11, 13], or after prolonged remission of their symptoms, become completely refractory to treatment [11]. Interestingly, several patients with HVDRR have shown spontaneous healing of their rickets [14-16]. This usually occurs between 7-15 years of age, sometimes after a period of ineffective long-term treatment with vitamin D and mineral replacement or after the treatment was discontinued. Spontaneous remission could explain the absence of radiological signs of active rickets in the first patient - 10 years old - at the first visit, and the spontaneous improvement of osteopenia and reduction of ALP in the second visit, while she was off therapy for the previous 13 months. Despite radiological improvement and being eucalcemic, she still had hyperparathyroidism, hypophosphatemia and elevated1,25(OH)2D3 level indicating that resistance to 1,25(OH)2D3 still exists. Perhaps after complete skeletal growth has been achieved her body was able to compensate for the defect in the VDR gene.
We detected a missense mutation in exon 2 in the first patient 22 (c.A 22 C) led to an altered amino acid at position 8 (p. Thr 8 Pro). This mutation is located in the N-terminal A / B regulatory domains containing the activation function 1 (AF-1) region. This A / B segment includes all residues that extend amino terminal to the DNA-binding domain (DBD) of the receptor. Its size is approximately 24 amino acids in the VDR [17]. Expected effect is weak ligand-independent transcription [1].
A second missense mutation was detected at initiation codon of exon 3 in patients, their father and healthy brother (c. A155G), leading to an altered amino acid at position 51 (p. Ser 51 Gly). Hsieh, et al. [18] mentioned that Ser-51 lies at the start of the second zinc finger that constitutes the DBD of VDR. Mutation in Ser-51 can greatly affect the transactivation ability of the VDR as Ser-51 phosphorylation is an important step in the control of VDR transcription [19].A number of mutations in the VDR-DBD had been reported previously [20, 21] but to our knowledge this is the first report that detects mutation of Ser-51in exon 3. Although mutations in DNA-binding domain is known to inactivate the VDR by disrupting contact with VDREs in promoters of target genes [22], and lead to severe hormone resistance and alopecia. Our patients did not have alopecia and responded well to oral treatment. Presence of DBD mutation in a heterozygous state results in less severe disease.
VDR-RFLP analysis of exons 7 and 8 revealed a heterozygous mutation at the GTAC sequence at the 970 bp affecting both patients and their mother. A C to A substitution mutation was previously reported by Malloy, et al. [23] in seven related families with HVDRR. The defect was conversion of the nucleotide sequence GTAC to GTAA at 970 bp at (exon 7) of VDR. This mutation results in an ochre nonsense mutation in the VDR, leading to a premature termination signal within the ligand binding domain of the VDR. The mutant protein was unable to bind with1,25(OH)2D3 and thus non functional [23].
In addition to our case, four other cases of HVDRR were shown to be caused by compound heterozygous mutations in the VDR. One French patient had two missense mutations in the VDR LBD that resulted in L263R and R391S amino acid substitutions. This patient responded to treatment with extremely high dose of vitamin D and calcium supplementation [24]. The second patient is a 55-year-old Caucasian woman with HVDRR that was resistant to treatment as a young child. She had a single base deletion in exon 4 that led to a frame shift and downstream termination codon in exon 4 and a missense mutation in the VDR LBD that resulted in an E329K amino acid substitution [25]. The third patient is a girl born to a father from the Czech Republic and a mother from Indonesia. She had a nonsense mutation R30X in the DBD that changed the arginine to stop codon at position 30 compound with a second unique 3-bpin-frame deletion in exon 6 that deleted the codon for lysine at position 246. She partially responded to oral calcium and calcitriol therapy [26]. All of these three patients had alopecia. The last patient is a Korean female with a 719 C-to-T (I146T) mutation in exon 4, and 754 C-to-T (R154C) mutation in exon 5 of the VDR gene that resulted in HVDRR without alopecia that responded well to oral vitamin D and calcium therapy [27].
Two previous study identified VDR gene mutation in 2 Saudi girls with HVDRR. The first one had a point mutation at nucleotide 252 in exon 2 of the VDR. This missense mutation (GGC to GAC) resulted in the conversion of glycine to aspartic acid at amino acid position 46 (G46D), located at the base of the first zinc finger [28]. The second girl had T to C mutation in exon 7 that changed isoleucine to threonine at amino acid 268 (I268T). The mutation resulted in HVDRR without alopecia that responded only to IV calcium infusion [29].
The presence of one allele of any of the detected mutation results in partial resistance to the1,25(OH)2D3 as shown by elevated PTH levels in both parents. This can also explain the associated hypophosphatemia in the mother. But if this is true, why do healthy heterozygous brother show normal biochemical results, and normal parathyroid hormone levels? It is possible that the activity of 1α hydroxylase enzyme at the younger age can compensate for the partial resistance to vitamin D action.
In conclusion we identified compound heterozygous mutation as the molecular basis for HVDRR in a patient with HVDRR without alopecia. This form responds easily to oral treatment with high dose vitamin D and calcium supplementation. The results of our study warrant further functional analysis to detect the effect of detected mutations on protein function, and their effect on the binding capacity of the vitamin D receptor.
| References | ▴Top |
- Malloy PJ, Pike JW, Feldman D. The vitamin D receptor and the syndrome of hereditary 1,25-dihydroxyvitamin D-resistant rickets. Endocr Rev. 1999;20(2):156-188.
pubmed doi - Malloy PJ, Feldman D. Hereditary 1,25 dihydroxyvitamin D resistant rickets. In: Feldman D, Pike JW, Glorieux F, eds. Vitamin D 2. San Diego: Elsevier, 2005. 1207–1238.
- Feldman D, Chen T, Cone C, Hirst M, Shani S, Benderli A, Hochberg Z. Vitamin D resistant rickets with alopecia: cultured skin fibroblasts exhibit defective cytoplasmic receptors and unresponsiveness to 1,25(OH)2D3. J Clin Endocrinol Metab. 1982;55(5):1020-1022.
pubmed doi - Hewison M, Rut AR, Kristjansson K, Walker RE, Dillon MJ, Hughes MR, O'Riordan JL. Tissue resistance to 1,25-dihydroxyvitamin D without a mutation of the vitamin D receptor gene. Clin Endocrinol (Oxf). 1993;39(6):663-670.
pubmed doi - Marx SJ, Liberman UA, Eil C, Gamblin GT, DeGrange DA, Balsan S. Hereditary resistance to 1,25-dihydroxyvitamin D. Recent Prog Horm Res. 1984;40:589-620.
pubmed - Liberman UA, Marx SJ. Vitamin D-dependent rickets. In: Favus MJ, ed. Primer on the metabolic bone diseases and disorders of mineral metabolism. 5th ed. Washington DC: American Society for Bone and Mineral Research. 2003:407–413.
- Baker AR, McDonnell DP, Hughes M, Crisp TM, Mangelsdorf DJ, Haussler MR, Pike JW, et al. Cloning and expression of full-length cDNA encoding human vitamin D receptor. Proc Natl Acad Sci U S A. 1988;85(10):3294-3298.
pubmed doi - Liberman UA. Vitamin D-resistant diseases. J Bone Miner Res. 2007;22 Suppl 2:V105-107.
pubmed - Hughes MR, Malloy PJ, Kieback DG, Kesterson RA, Pike JW, Feldman D, O'Malley BW. Point mutations in the human vitamin D receptor gene associated with hypocalcemic rickets. Science. 1988;242(4886):1702-1705.
pubmed doi - Brooks MH, Bell NH, Love L, Stern PH, Orfei E, Queener SF, Hamstra AJ, et al. Vitamin-D-dependent rickets type II. Resistance of target organs to 1,25-dihydroxyvitamin D. N Engl J Med. 1978;298(18):996-999.
pubmed doi - Balsan S, Garabedian M, Liberman UA, Eil C, Bourdeau A, Guillozo H, Grimberg R, et al. Rickets and alopecia with resistance to 1,25-dihydroxyvitamin D: two different clinical courses with two different cellular defects. J Clin Endocrinol Metab. 1983;57(4):803-811.
pubmed doi - Liberman UA, Samuel R, Halabe A, Kauli R, Edelstein S, Weisman Y, Papapoulos SE, et al.End-organ resistance to 1,25-dihydroxycholecalciferol. Lancet. 1980;1(8167):504-506.
pubmed doi - Beer S, Tieder M, Kohelet D, Liberman OA, Vure E, Bar-Joseph G, Gabizon D, et al. Vitamin D resistant rickets with alopecia: a form of end organ resistance to 1,25 dihydroxy vitamin D. Clin Endocrinol (Oxf). 1981;14(4):395-402.
pubmed doi - Hochberg Z, Benderli A, Levy J, Vardi P, Weisman Y, Chen T, Feldman D. 1,25-Dihydroxyvitamin D resistance, rickets, and alopecia. Am J Med. 1984;77(5):805-811.
pubmed doi - Hirst MA, Hochman HI, Feldman D. Vitamin D resistance and alopecia: a kindred with normal 1,25-dihydroxyvitamin D binding, but decreased receptor affinity for deoxyribonucleic acid. J Clin Endocrinol Metab. 1985;60(3):490-495.
pubmed doi - Chen TL, Hirst MA, Cone CM, Hochberg Z, Tietze HU, Feldman D. 1,25-dihydroxyvitamin D resistance, rickets, and alopecia: analysis of receptors and bioresponse in cultured fibroblasts from patients and parents. J Clin Endocrinol Metab. 1984;59(3):383-388.
pubmed doi - Miyamoto K, Kesterson RA, Yamamoto H, Taketani Y, Nishiwaki E, Tatsumi S, Inoue Y, et al. Structural organization of the human vitamin D receptor chromosomal gene and its promoter. Mol Endocrinol. 1997;11(8):1165-1179.
pubmed doi - Hsieh JC, Jurutka PW, Galligan MA, Terpening CM, Haussler CA, Samuels DS, Shimizu Y, et al. Human vitamin D receptor is selectively phosphorylated by protein kinase C on serine 51, a residue crucial to its trans-activation function. Proc Natl Acad Sci U S A. 1991;88(20):9315-9319.
pubmed doi - Nguyen TM, Adiceam P, Kottler ML, Guillozo H, Rizk-Rabin M, Brouillard F, Lagier P, et al. Tryptophan missense mutation in the ligand-binding domain of the vitamin D receptor causes severe resistance to 1,25-dihydroxyvitamin D. J Bone Miner Res. 2002;17(9):1728-1737.
pubmed doi - Sone T, Marx SJ, Liberman UA, Pike JW. A unique point mutation in the human vitamin D receptor chromosomal gene confers hereditary resistance to 1,25-dihydroxyvitamin D3. Mol Endocrinol. 1990;4(4):623-631.
pubmed doi - Saijo T, Ito M, Takeda E, Huq AH, Naito E, Yokota I, Sone T, et al. A unique mutation in the vitamin D receptor gene in three Japanese patients with vitamin D-dependent rickets type II: utility of single-strand conformation polymorphism analysis for heterozygous carrier detection. Am J Hum Genet. 1991;49(3):668-673.
pubmed - Malloy PJ, Feldman D. A novel mutation in the deoxyribonucleic acid-binding domain of the vitamin D receptor causes hereditary 1,25-dihydroxyvitamin D-resistant rickets. In Holick MF, ed. Vitamin D physiology, molecular biology, and clinical application. Part 4, 2nd ed. Totowa, NJ, Springers 2010:691-714.
- Malloy PJ, Hochberg Z, Tiosano D, Pike JW, Hughes MR, Feldman D. The molecular basis of hereditary 1,25-dihydroxyvitamin D3 resistant rickets in seven related families. J Clin Invest. 1990;86(6):2071-2079.
pubmed doi - Nguyen M, d'Alesio A, Pascussi JM, Kumar R, Griffin MD, Dong X, Guillozo H, et al. Vitamin D-resistant rickets and type 1 diabetes in a child with compound heterozygous mutations of the vitamin D receptor (L263R and R391S): dissociated responses of the CYP-24 and rel-B promoters to 1,25-dihydroxyvitamin D3. J Bone Miner Res. 2006;21(6):886-894.
pubmed doi - Miller J, Djabali K, Chen T, Liu Y, Ioffreda M, Lyle S, Christiano AM, et al. Atrichia caused by mutations in the vitamin D receptor gene is a phenocopy of generalized atrichia caused by mutations in the hairless gene. J Invest Dermatol. 2001;117(3):612-617.
pubmed doi - Zhou Y, Wang J, Malloy PJ, Dolezel Z, Feldman D. Compound heterozygous mutations in the vitamin D receptor in a patient with hereditary 1,25-dihydroxyvitamin D-resistant rickets with alopecia. J Bone Miner Res. 2009;24(4):643-651.
pubmed doi - Song JK, Yoon KS, Shim KS, Bae CW. Novel compound heterozygous mutations in the vitamin D receptor gene in a Korean girl with hereditary vitamin D resistant rickets. J Korean Med Sci. 2011;26(8):1111-1114.
pubmed doi - Lin NU, Malloy PJ, Sakati N, al-Ashwal A, Feldman D. A novel mutation in the deoxyribonucleic acid-binding domain of the vitamin D receptor causes hereditary 1,25-dihydroxyvitamin D-resistant rickets. J Clin Endocrinol Metab. 1996;81(7):2564-2569.
pubmed doi - Malloy PJ, Xu R, Peng L, Peleg S, Al-Ashwal A, Feldman D. Hereditary 1,25-dihydroxyvitamin D resistant rickets due to a mutation causing multiple defects in vitamin D receptor function. Endocrinology. 2004;145(11):5106-5114.
pubmed doi
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.