

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website http://www.jofem.org |
Original Article
Volume 1, Number 3, August 2011, pages 125-141
Glycyrrhizic Acid Improves Lipid and Glucose Metabolism in High-Sucrose-Fed Rats
Chanchal Chandramoulia, Yong Sheau Tinga, Lam Yi Lyna, Ton So Haa, c, Khalid Abdul Kadirb
aSchool of Sciences, Monash University Sunway Campus, Jalan Lagoon, Selatan, Bandar Sunway 46150, Selangor Darul Ehsan, Malaysia
bSchool of Medicine and Health Sciences, Monash University Sunway Campus, Jalan Lagoon Selatan, Bandar Sunway 46150, Selangor Darul Ehsan, Malaysia
cCorresponding author: Ton So Ha
Manuscript accepted for publication August 22, 2011
Short title: GA in Lipid and Glucose Metabolism
doi: https://doi.org/10.4021/jem39w
| Abstract | ▴Top |
Background: The alarming increase in sugar consumption worldwide has largely contributed towards the escalating symptoms of metabolic syndrome (MetS) such as obesity, insulin resistance and dyslipidaemia. Reduction in lipoprotein lipase (LPL) activity leads to dyslipidaemia, a hallmark of MetS. Increased activation of glucocorticoid receptors also results in symptoms of MetS. Glycyrrhizic acid (GA), a triterpenoid saponin, inhibits 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1), which leads to reduced hexose-6-phosphate (H6PDH) activity and decreased glucocorticoids (GCs) production. GCs induce the gene transcriptions of enzymes such as phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) in the gluconeogenic pathway. Studies have indicated that triterpenoids could act as PPAR agonists and GA is therefore postulated to restore LPL expression in the insulin resistant state.
Methods: Twenty-four male Sparague Dawley rats were randomly divided into three treatment groups (Group A, normal diet; Group B, high-sucrose diet; Group C, high-sucrose diet with 100 mg/kg of GA). After 28 days of treatment, the blood samples collected were analyzed for blood glucose, serum insulin and lipid profiles and the tissues collected were used for LPL expression, 11β-HSD1, H6PDH and PEPCK activities analysis. Statistical analysis was done using REST9 and SPSS softwares, P ≤ 0.05 was considered significant.
Results: Oral administration of 100mg/kg GA to high-sucrose induced rats significantly lower blood glucose (32.2%), serum insulin (68.4%), HOMA-IR (81.6%) (P < 0.05) and improved lipid parameters (P < 0.05) with no elevations in blood pressure. LPL activity was upregulated in all tissues (P > 0.05), with significant upregulation in the liver (2 fold) (P < 0.01). 11ß-HSD activity was lower in all tissues in GA treated rats compared to the group on high-sucrose diet alone (P < 0.05). Smaller elevations in H6PDH activities were observed in the liver (14.2%), kidney (7.17%), subcutaneous adipose tissue (SAT)(15.3%), abdominal muscle (AM) (18.4%) and quadriceps femoris (QF) (15.3%). PEPCK activities were elevated in the liver (14.9%) and kidney (7.0%) (P < 0.05) but increased in the SAT (74.4%) and visceral adipose tissues (VAT) (142.0%) (P < 0.01). Smaller increases were observed in the hepatic (61.6%) and renal (47.4%) G6Pase activities (P < 0.05).
Conclusion: High-sucrose feeding induced hypertension, hyperglycemia, insulin resistance and dyslipidemia. GA could ameliorate these symptoms by regulating the glucose and lipid metabolism through 11β-HSD1 inhibition and possibly PPARγ agonism. GA induced LPL expression and contributed towards a postive shift in the lipid profiles. GA also prevented hyperglycaemia and improved insulin sensitivity in the high-sucrose fed rats without causing hypertension. Thus, GA appears to be a possible therapeutic compound in lowering the risks of developing dyslipidaemia and possibly T2DM.
Keywords: Glycyrrhizic acid; Dyslipidaemia; Diabetes; Lipoprotein lipase (LPL); Phosphoenolpyruvate Carboxykinase (PEPCK); Hexose-6-Phosphate Dehydrogenase (H6PDH); Glucose-6-Phosphatase (G6Pase); 1β-hydroxysteroid dehydrogenase 1 (11 β-HSD)
| Introduction | ▴Top |
The metabolic syndrome (MetS), also known as the insulin resistance syndrome and syndrome X, refers to a clustering of several risk factors for atherosclerotic cardiovascular disease (CVD) and Type 2 Diabetes Mellitus (T2DM) [1-3]. High-sucrose diet is often implicated with symptoms of MetS which include abdominal obesity, hypertension, insulin resistance and hypertriglyceridemia. Glucocorticoids (GCs) play important roles in glucose and lipid metabolism [4]. High-sucrose feeding may have an impact on GC metabolism via their effect on 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1) activity. This results in an increase in active GCs which lead to insulin resistance (IR) and T2DM [5].
Various studies have shown that with the development of IR, the production and activity of the enzyme lipoprotein lipase (LPL) is reduced [6]. LPL is a central enzyme involved in lipid metabolism. LPL hydrolyzes the core triacylglycerol (TAG) of circulating TAG-rich lipoproteins and facilitate the entry of fatty acids into the peripheral tissues. LPL is regulated by the upstream peroxisome proliferator activator receptor (PPAR) [7]. Reduced LPL activity has been shown to lead to a reduction in the lipolytic rate of the chylomicrons and the very-low-density lipoproteins (VLDL). This results in gradual development of hypertriglyceridaemia and subsequently dyslipidaemia [8].
Insight on the role of LPL comes from different animal models where LPL has either been removed using targeted gene knockout or over-expressed in specific tissues in transgenic mouse lines. Within 18 hours of birth, homozygous knockout animals showed an 80-fold increase in plasma TAG and subsequently failed to survive [9]. This lethal homozygous phenotype could be ‘rescued’ by muscle-specific expression of LPL [10]. Adult heterozygous LPL knockout mice showed delayed clearance of triglyceride-rich-lipoproteins (TRL). However, overexpression of LPL gene in the skeletal muscle and liver has been demonstrated to lead to IR owing to the accumulation of TAG stores which fails to enter the hydrolytic pathway [11]. Hence tissue specific regulation of LPL is important for proper fuel partitioning, body weight regulation and insulin action.
Overexpression of hexose-6-phosphate dehydrogenase (H6PDH), phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) were observed in T2DM patients when GCs were found to be in excess [12, 13]. H6PDH catalyzes the first two steps of the pentose phosphate pathway within the endoplasmic reticulum lumen. It has a bifunctional activity where H6PDH oxidizes glucose-6-phosphate (G6P) to 6-phosphogluconolactone (6PGL) and concomitantly generates reduced nicotinamide adenine dinucleotide phosphate (NADPH) from NADP+ [14, 15]. The conversion of inactive GCs to active GCs is catalyzed by 11β-HSD1, which utilizes NADPH generated from H6PDH [16]. Thus, the reductase activity of 11β-HSD1 is strongly dependent on the availability of NADPH, which further relies on G6P availability within the ER lumen [14].
GCs, as insulin antagonists, were found to inhibit the release of insulin by the pancreatic β cells in mice and disrupt the high-frequency insulin secretion in the fasting state in human [17]. They promote free fatty acid (FFA) synthesis and stimulate the release of lipoproteins in the liver. This leads to fat accumulation in hepatocytes which may impair the insulin-induced inhibition of gluconeogenesis [18]. Moreover, GCs stimulate hepatic gluconeogenesis through the activation of glucocorticoid receptor (GR), which upregulates the expression of PEPCK and G6Pase, the two key enzymes involved in gluconeogenesis. They also suppress the insulin-dependent glucose uptake and inhibit glycogen synthesis, resulting in reduced insulin sensitivity and elevated hepatic glucose production. These contribute to the development of hyperglycaemia, a characteristic of IR [19].
Currently the available classes of drugs against dyslipidaemia include the statins, fibrates and the NO-1886 (ibrolipim). While statins act onhydroxymethylglutaryl-CoA reductase (HMG Co-A) to lower cholesterol levels, fibrates function as PPAR agonists to induce the increase in LPL activity [20].On the other hand, the antidiabetic drugs available in the market include sulfonylureas, metformin and thiazolidinediones (TZDs) [21]. TZDs promote peripheral glucose uptake and improve insulin sensitivity in the skeletal muscle, liver and adipose tissue [22]. Likewise, metformin being a biguanide agent inhibits hepatic glucose production and enhances peripheral glucose uptake, leading to amelioration of hyperglycaemia. It has been demonstrated to stimulate a cascade of reactions, which in turn suppresses gluconeogenesis through inhibition of PEPCK and G6Pase gene expressions [23].
However, these drugs are associated with several drawbacks. Despite the effectiveness of statins and fibrates in hypercholestrolaemic patients, long-term consumption of these drugs result in muscle fatigue and increased incidences of cholesterol gallstone respectively [24]. The TZDs on the other hand is associated with fluid retention and plasma volume expansion which lead to peripheral oedema. This increases the incidence of heart failure in some patients, with a frequency of 2.5 times greater in patients on combination therapy with insulin [25]. On the other hand, NO-1886 has been reported to inhibit basal and adrenocorticotrophic hormone (ACTH)-induced release of steroid hormones in rat, dog, monkey and human adrenocortical cells, resulting in hypertrophy of the adrenal glands [26]. Metformin is also associated with undesirable side effects such as lactic acidosis, renal failure and other adverse effects such as abdominal discomfort and diarrhoea [21]. It is therefore crucial to find an alternative LPL-raising therapeutic agent and an anti-diabetic drug to ameliorate dyslipidaemia and T2DM.
Glycyrrhizic acid (GA), a triterpenoid saponin, is the primary bioactive constituent of the roots of the shrub Glycyrrhiza glabra. GA and its active metabolite glycyrrhetic acid act as a potent inhibitor of 11β-HSD1 [27]. Suppression of 11β-HSD1 favors the conversion of active GC to its inactive form, resulting in reduced GCs production [28]. Such manipulations of GC action can be targeted as a therapeutic strategy for the treatment of T2DM [29]. More importantly, triterpenoids have been known to act as PPAR agonists [30, 31]. This may suggest that GA could also potentially act to activate the PPAR class of nuclear receptors and may therefore be proposed to be a candidate for raising LPL. Thus, GA may be an anti-dyslipidaemic and anti-diabetic drug.
Thus in the present study the role of GA in glucose and lipid metabolism was investigated by determining blood pressure, blood glucose, serum insulin, Homeostatic Model Assessment for Insulin Resistance (HOMA-IR), TAG, FFA, total cholesterol, LDL, HDL, LPL expression and activities of PEPCK, H6PDH, G6Pase and 11β-HSD1.
| Materials and Methods | ▴Top |
Animal treatment
The use and handling procedure of animals in this research project had been approved by the Monash University School of Biomedical Sciences Animal Ethics Committee (AEC Approval Number: SOBSB/MY/2010/43). Twenty-four male Sprague Dawley rats (Rattus novergicus) weighing between 200 - 250 g were obtained from the Animal House of Monash University Sunway Campus and were housed individually in polypropylene cages in a room kept at 23 °Con a 12-hour light and dark cycle (lights on at 0600 hours and lights out at 1800 hours). These rats were randomly segregated into three groups of eight, namely groups A, B and C. Rats from group A were fed on a normal diet with 25 g of standard rat chow and water. Rats from group B were fed on 25 g of high-sucrose pellets and water. The high-sucrose diet pellets were prepared by mixing powdered rat chow to cane sugar in the ratio of 2:3. Rats from group C were also fed on high-sucrose pellets but were orally given 100 mg/kg of GA. Throughout the treatment period of 28 days, daily food and fluid consumption were monitored.
Systolic blood pressure
The NIBP controller (ADInstruments, Australia) was used to measure the systolic blood pressure of the rats. Conscious rats were placed into a plastic restrainer of which the length was adjusted to fit the animal comfortably while their movements restricted. A tail-cuff with a pulse transducer was applied onto the tail of the restrained animal and the tail was heated for 20 - 30 minutes using a table lamp with 60W bulb in order to dilate the caudal arteries. Blood pressure was measured by inflating the tail-cuff once the heart rate had stabilised. The recording and determination of blood pressure were performed using the Chart recording software and a final reading was averaged out from at least ten consecutive readings.
Sample collection
At the end of the treatment period, the rats were fasted for 12 hours prior to humane sacrifice between 0800 to 1000 hours under the influence of anaesthesia via intraperitoneal administration of pentobarbital sodium (Nembutal) (150 mg/kg). Blood was drawn from the cardiac ventricle via the apex and was centrifuged at 12,000 × g for 10 minutes. The resulting serum supernatant was then rapidly aliquoted into microtubes and kept frozen at -80 °Cuntil required for analysis. The seven tissues of interest: liver (L), kidney (K), heart (H), abdominal muscle (AM), quadriceps femoris (QF) and visceral (VAT) and subcutaneous adipose tissues (SAT) were harvested immediately following dissection. Fractions of these tissues were placed in individual cryovials (Nalgene, USA) and immediately flash-frozen in liquid nitrogen. These fractions were then stored at -80 °C until required for RNA analysis. Six tissues which included L, K, AM, QF, SAT and VAT were also individually harvested and placed in Krebs-Ringer bicarbonate (KRB) buffer (pH 7.4) for 11ß-HSD activity determination. For PEPCK determination, the liver, kidneys, SAT and VAT were placed into falcon tubes containing 5mL of Tris-HCl buffer, pH 7.4 at 4 °C. For H6PDH and G6Pase determinations, the liver, kidneys, SAT, VAT, AM and QF tissues were placed into tubes containing HEPES-KOH buffer, pH 7.2 at 4 °C. Further details on tissue collection and processing are detailed in the respective sections.
Blood and serum biochemical analysis
Blood glucose was determined using Trinder’s glucose oxidase method while serum insulin was determined using the Rat/Mouse Insulin ELISA Kit (Linco Research, USA). The HOMA-IR was calculated as the product of fasting blood glucose and serum insulin divided by 22.5 as described by Alberts et al [32].Serum FFA, TAG and total cholesterol were determined using the Randox FA115 Non-Esterified Fatty Acids kit (Randox, UK), Randox Triglycerides Kit (Randox, UK) and Randox CH200 Cholesterol Kit (Randox, UK) respectively. HDL-cholesterol was initially separated from the LDL and VLDL fraction by precipitation of the latter using the Randox CH203 HDL Precipitant (Randox, UK). HDL-cholesterol was then determined using the aforementioned cholesterol kit. LDL-cholesterol was calculated using the levels of total cholesterol, TAG, HDL cholesterol obtained using the Friedewald formula [33].
Real time reverse transcription polymerase chain reaction (qRT-PCR) of lipoprotien lipase (LPL) gene
Total RNA was isolated from the liver, kidney, AM and QF using the Qiagen RNeasy Mini kit (Qiagen, USA) while Qiagen RNeasy Lipid Tissue Mini kit was used to isolate the total RNA from adipose tissues, SAT and VAT (Qiagen, USA). The RNA purity was determined by measuring the absorbance of the diluted RNA at 260 and 280 nm. RNase-free DNase treatment was performed using Promega RQ1 RNase-free DNase (Promega, USA) and cDNA synthesis was performed using the Qiagen Omniscript Reverse Transcriptase kit (Qiagen, USA). Relative LPL expression was determined by qT-PCR using the Comparative Ct (ΔΔCt) Method, normalized to the BAC gene. Sequence of primers and probes specific to Rattus norvegicus LPL [GenBank: BC081836] and BAC [Gen- Bank: BC063166] mRNA are as follows:
LPL, forward: 5'-CAGCAAGGCATACAGGTG-3'
LPL, reverse: 5'-CGAGTCTTCAGGTACATCTTAC-3'
LPL, probe: 5'-(6-FAM) TTCTCTTGGCTCTGACC(BHQ1)-3'
BAC, forward: 5'-GTATGGGTCAGAAGGACTCC-3'
BAC, reverse: 5'-GTTCAATGGGGTACTTCAGG-3'
BAC, probe: 5'-(TET) CCTCTCTTGCTCTGGGC(BHQ1)-3'
Agarose gel electrophoresis was performed on amplicons produced from qRT-PCR to assess primer specificity. LPL gene expression in different tissues was determined using the 2-ΔΔCt calculation. The values of fold differences were obtained using BAC as the endogenous reference gene and the change in threshold value (ΔCt) between target and calibrator depending on the type of comparison made. Two types of comparison were carried out, i.e. (i) Group A as the calibrator and Group B as the target; (ii) Group B as the calibrator and Group C as the target. Therefore, LPL expression of all tissues in the control group was given a base value of 1.00 [34].
Protein determination
Prior to enzyme activity measurement, samples were homogenized using Heidolph DIAX 900 rotor stator homogenizer (Sigma-Aldrich, U.S.A) and centrifuged to obtain appropriate fractions. The protein concentrations of collected samples were determined using a modified Lowry’s method (Bio-Rad DC Protein Assay).
Glucocorticoid (GC) quantification using high performance liquid chromatography (HPLC)
A total of 50mg of protein was added into substrate mixtures of 11β-HSD 1 and an excess of cofactors: 0.35mM NADP+ for 11β-HSD Type 1, 0.2% BSA, 0.2% glucose and 5% ethanol. A total of 800 µl ethyl acetate was added to the tubes and placed horizontally on an orbital shaker rotating at 100 rpm for 30 minutes at room temperature (25 °C). Upon centrifugation at 16,000 g for 10 mins , the ethyl acetate was evaporated under the flow of nitrogen gas. Reverse-phase HPLC chromatography was used to quantify the glucocorticoid, 11-dehydrocorticosterone, which is the product of the dehydrogenation of the substrate, corticosterone. The mobile phase consisted of 20% methanol, 30% acetonitrile and 50% water (v/v). The dried protein contents wereresuspended in 300 µl high performance liquid chromatography mobile phase. The glucocorticoids were separated via reverse phase chromatography using Waters Symmetry® C18 column (3.9 mm x 150 mm ) by a linear methanol-acetonitrile-water gradient from 10:15:75 (v/v) to 20:30:50 (v/v) in the first five minutes followed by isocratic elution in the next five minutes (Perkin Elmer 200 Liquid Chromatography). The Diode Array Detector was used to measure the absorbance of the glucocorticoids at 254 nm. The concentration of 11-dehydrocorticosterone in the samples was determined by referring to the absorbance values (expressed as area under curve) from the 11-dehydrocorticosterone standard curve. 1U of 11β-HSD activity was defined as one picomole of 11-dehydrocorticosterone produced/50mg of tissue protein used/hour.
Phosphoenolpyruvate carboxykinase (PEPCK) activity
PEPCK activities in the liver, kidneys, SAT and VAT were determined according to a protocol by Petrescu et al [35]. A total of 80 µL of standards and samples were added in duplicates into a 96-well microtitre plate. Each well contained a reaction mixture consisting of 50 mM Tris-HCl (pH 7.4), 50 mM NaHCO3, 1 mM MnCl2, 1mM phosphoenolpyruvate, 2U malate dehydrogenase and 0.25 mM NADH. The plate was incubated at 30 °C for 3 minutes. The reaction was initiated upon the subsequent addition of 0.15 mM 2’-deoxyguanosine 5’-diphosphate (dGDP). The spectrometric absorbance was measured at 340 nm at every one minute interval for 5 minutes. One unit of PEPCK activity was expressed as nmoles NADH oxidized/min/mg protein.
Hexose-6-Phosphate dehydrogenase (H6PDH) activity
H6PDH assay was carried out as described by Banhegyi et al [36]. The microsomal fractions of the liver, kidneys, SAT, VAT, AM and QF were permeabilized with 10% Triton X-100 prior to incubation at 22 °C for 3 minutes. A series of NADPH standards ranging from 0.05 mM to 0.25 mM were prepared. A total of160 µL of standards and permeabilized samples were added in duplicates into a 96-well microtitre plate. Each well contained a reaction mixture of 100 µMNADP+ and 10 µM glucose-6-phosphate (G6P). The spectrometric absorbance was measured at 340 nm at every one minute interval for 5 minutes. One unit of H6PDH activity was expressed as nmoles NADPH formed/min/mg protein.
Glucose-6-Phosphatase (G6Pase) activity
Determination of microsomal G6Pase activities in the liver and kidneys was carried out according to a protocol by Csala et al [37]. The microsomes were permeabilized with alamethicin (0.1 mg/mg microsomal protein). A 2.5 µL of the permeabilized samples was added to the wells of the microtitre plate prior to the addition of 5mM G6P and MOPS-KCl buffer. The reaction mixture was incubated at 37 °C for 2 minutes. The enzymatic reaction was terminated by adding stopping reagent (3.4 mM ammonium molybdate in 0.5M sulphuric acid, 0.52M sodium dodecyl sulphate (SDS) and 0.6 M ascorbic acid in the proportions of 6:2:1). The reaction mixture was further incubated at 45 °C for 20 minutes. The absorbance was measured at 820 nm. One unit of G6Pase activity was expressed as nmoles of inorganic phosphate produced/minute/milligram protein.
Statistical analysis
Statistical analysis for LPL expression was performed using the Relative Expression Software Tool (REST©) MCS Beta 2006. All other data were analyzed using Statistics Package for the Social Sciences (SPSS) for Windows Version 16.0. Data distribution was analyzed using the Kolmogorov-Smirnov normality test. Data with parametric distribution were then analyzed using Analysis of Variance (ANOVA) and if the results were significant, a Post Hoc Scheffe test was performed. Data with nonparametric distribution was analyzed using Kruskall-Wallis test. All data in this study were found to be parametric and hence were all reported as mean ± standard error. A P-value of ≤ 0.05 was considered to be statistically significant.
| Results | ▴Top |
Systolic blood pressure elevated with high-sucrose feeding
Throughout the four week treatment period, the mean systolic blood pressure of Groups B and C were consistently higher than Group A. The mean blood pressure was significantly elevated in Group B and Group C (110.63 ± 3.47 and 112.33 ± 2.06 mmHg) compared to Group A which recorded 101.65 ± 3.74 mmHg (P < 0.05) in Week 4. The difference between Groups B and C however was not significant throughout the four weeks (P > 0.05) (Fig. 1)
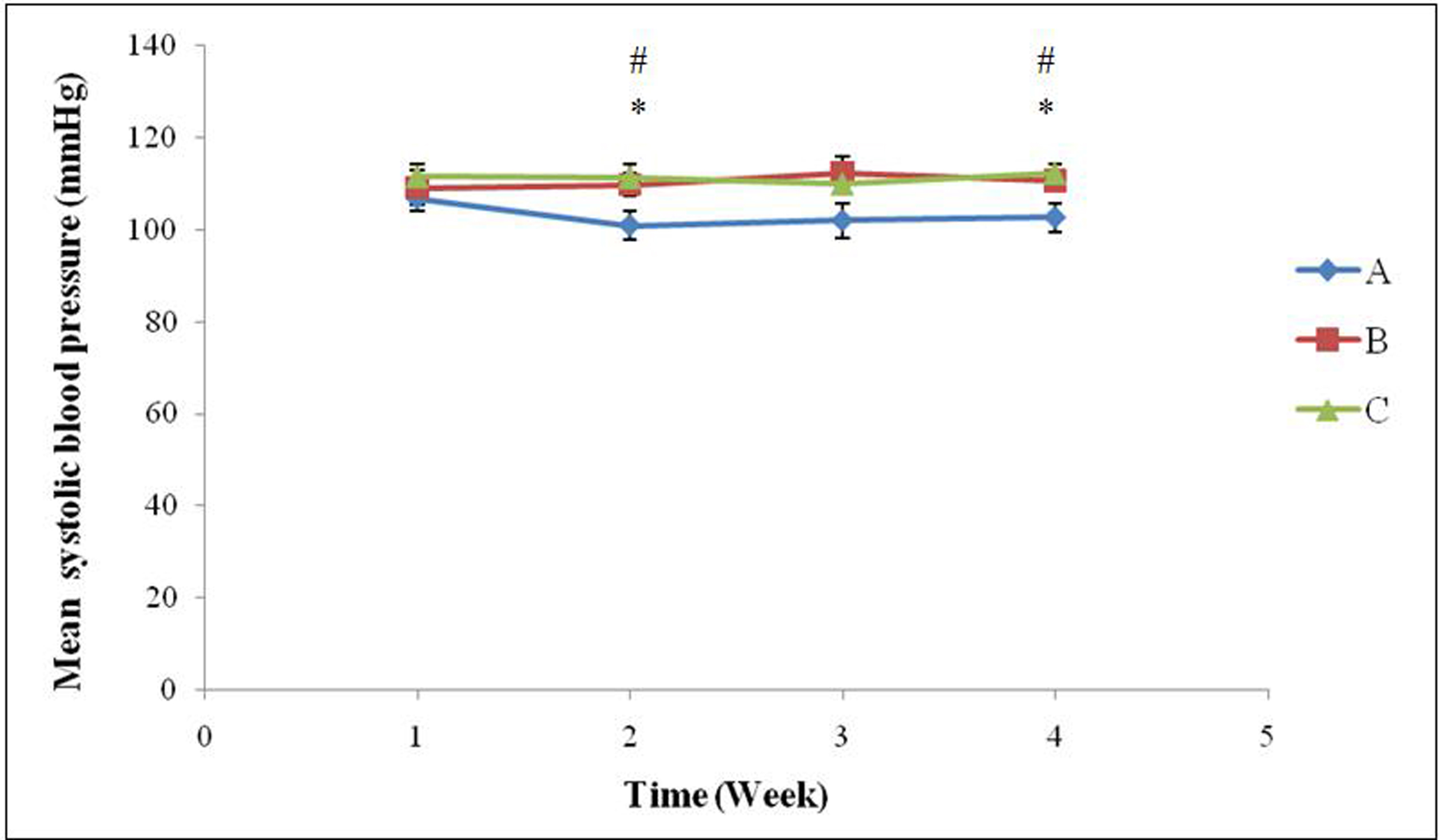 Click for large image | Figure 1. Mean systolic blood pressure (mmHg) in the three groups of rats over a 4-week treatment period (* indicates significant difference between Groups A and B (P < 0.05) and # indicates significant difference between Groups A and C (P < 0.05). Group A, Normal Diet; Group B, High-sucrose diet; Group C, High-sucrose diet + GA. |
GA treatment led to improved insulin sensitivity in high-sucrose fed rats
The mean blood glucose levels in groups A, B and C were 5.67 ± 0.15, 8.35 ± 0.52 and 5.66 ± 0.33 mM respectively. Group B had a significant increase (47.3%) compared to group A (P = 0.000, P < 0.01). There was no significant difference between groups A and C (P = 0.969, P > 0.05). A significant difference was observed between groups B and C (P = 0.000, P < 0.01), where the mean blood glucose level in group B was 47.5% higher than that in group C (Fig. 2).
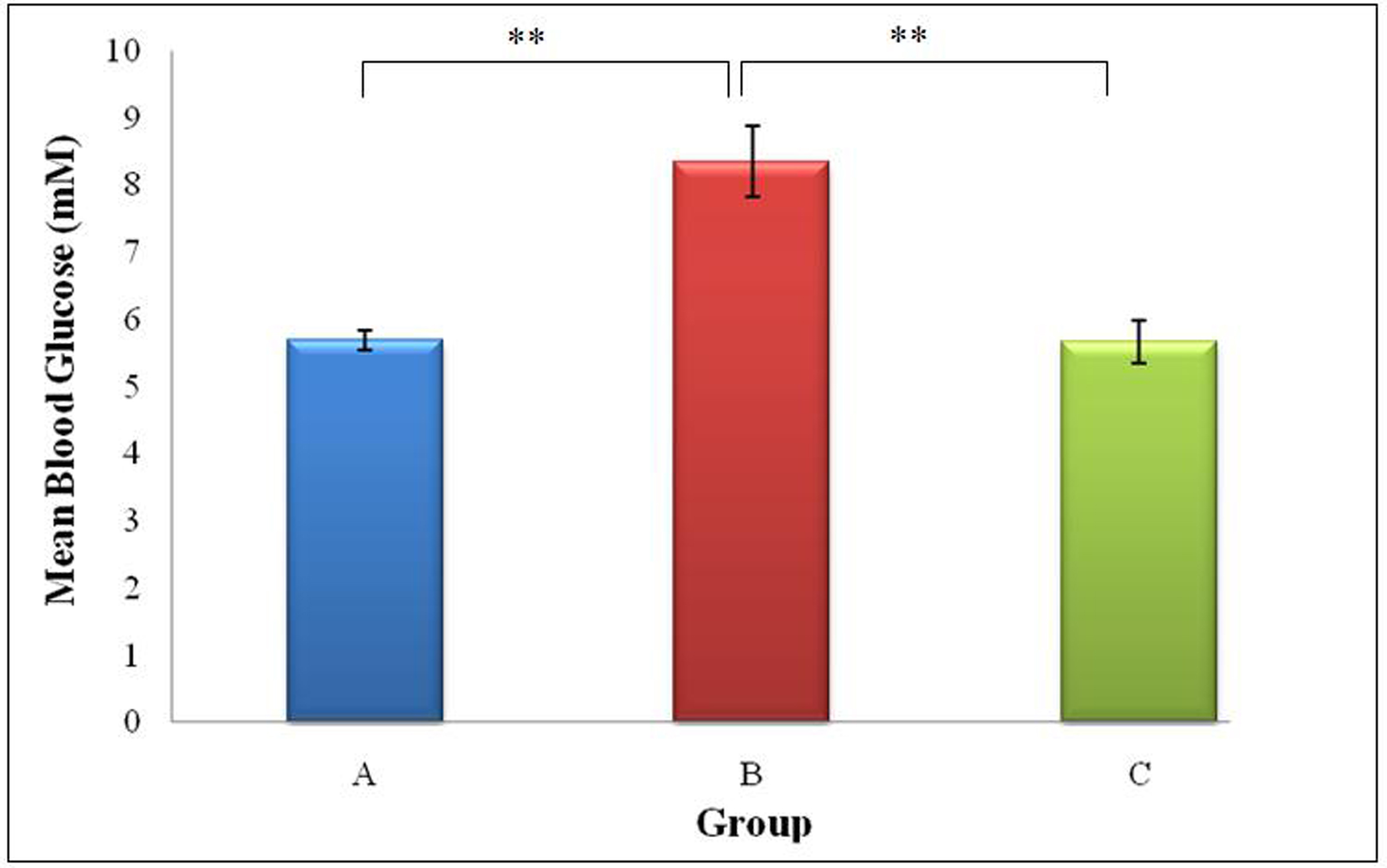 Click for large image | Figure 2. Mean blood glucose concentration (mmol/L) of rats from Groups A, B and C (** indicates P < 0.01 between groups).Group A, Normal Diet; Group B, High-sucrose diet; Group C, High-sucrose diet + GA. |
The mean serum insulin level of groups A, B and C were 0.08 ± 0.02 ng/ml, 0.95 ± 0.25 ng/ml and 0.28 ± 0.08 ng/ml respectively .The highest serum insulin level was observed in Group B. There was a significant 11.7 fold increase (P < 0.01) in Group B compared to Group A. However, the significant increaseobserved in Group C was moderated to a much lower 3.47 fold increase (P < 0.05). The difference between Groups B and C was also significant (3.39 fold, P < 0.05) (Fig. 3).
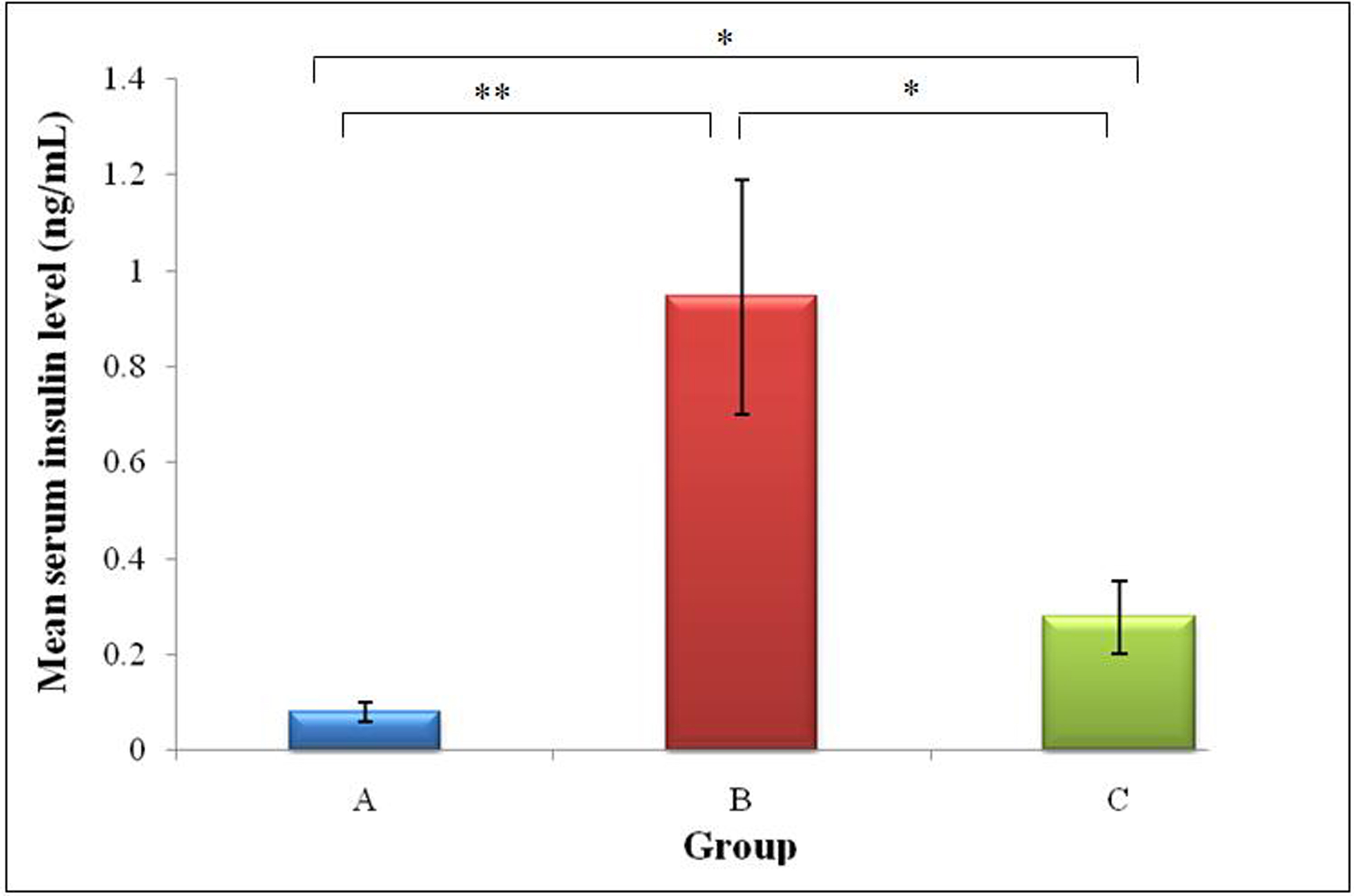 Click for large image | Figure 3. Mean serum insulin (ng/mL) of rats from Groups A, B and C (* indicates P < 0.05 and ** indicates P < 0.01 between groups). Group A, Normal Diet; Group B, High-sucrose diet; Group C, High-sucrose diet + GA. |
The HOMA-IR value was the highest in Group B (0.38 ± 0.09), followed by Group C (0.07 ± 0.02) and Group A (0.02 ± 0.01). With high-sucrose feeding, a 17.2-fold increase in HOMA-IR index was observed in Group B (P < 0.01) whereas, only a 3.16 -fold increase was observed in Group C compared to Group A (P < 0.05). The 5.44 -fold difference between Groups B and C was also significant (P < 0.01). The lower HOMA-IR in Groups A and C indicated higher insulin sensitivity (lower insulin resistance) in these groups compared to Group B. (Fig. 4).
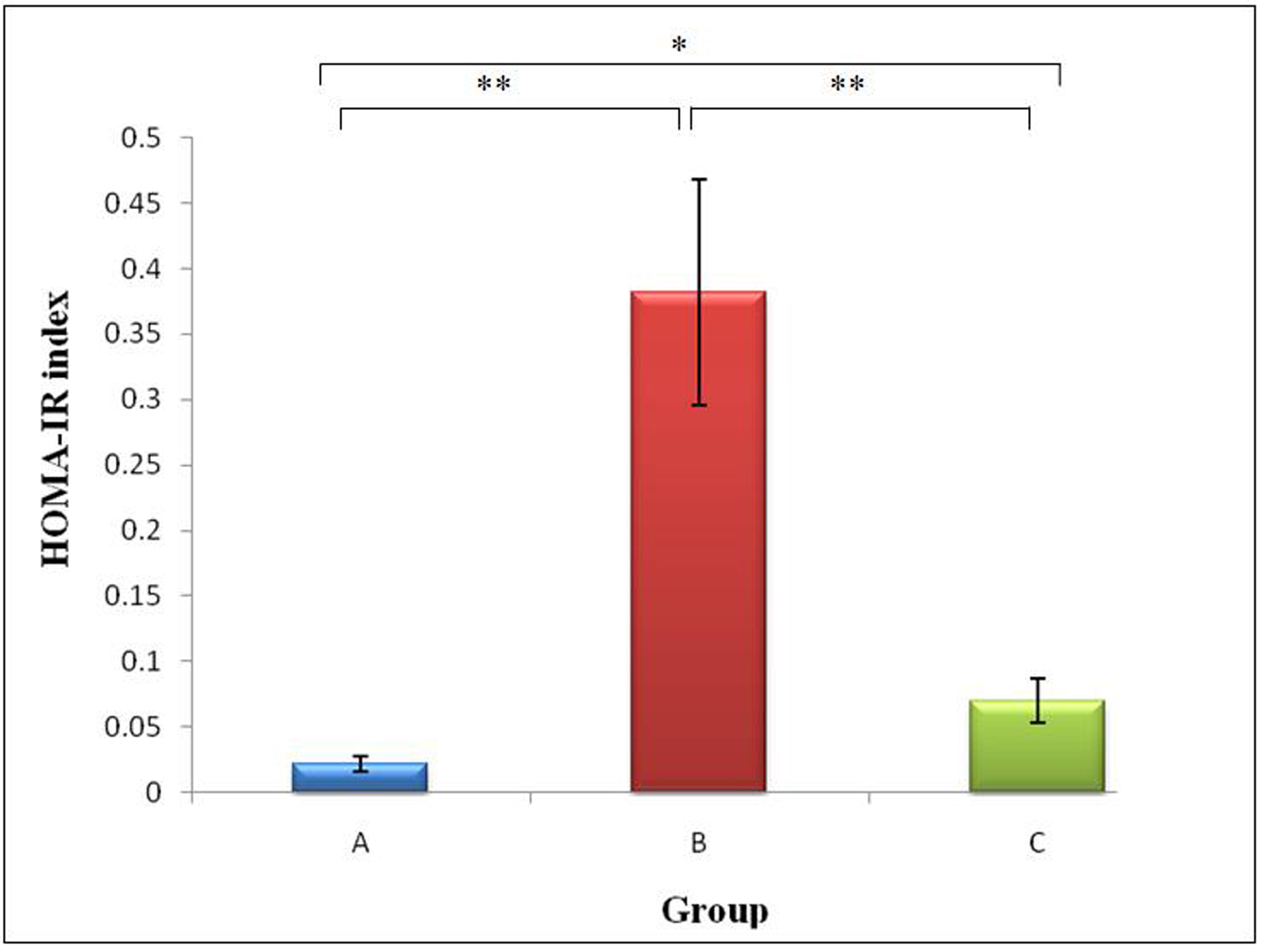 Click for large image | Figure 4. Mean HOMA-IR of rats from Groups A, B and C (* indicates P < 0.05 and ** indicates P < 0.01 between groups).Group A, Normal Diet; Group B, High-sucrose diet; Group C, High-sucrose diet + GA. |
GA administration resulted in a positive shift in lipid profiles of sucrose induced obese rats
A comparison of the lipid parameters and free fatty acids (FFA) between Groups A, B and C is shown in Figure 5. The mean serum FFA level of Groups A, B and C were 0.53 ± 0.03 mmol/L, 0.59 ± 0.07 mmol/L and 0.53 ± 0.05 mmol/L respectively. An increase was observed in Group B (11.3%), (P = 0.613, P > 0.05). GA prevented this increase in addition to potentiating a decrease of 0.2% in Group C compared to Group A (P = 0.493, P > 0.05). A difference of 10.16% was observed between Groups B and C (P = 0.855, P > 0.05). However, none of these results were significant.
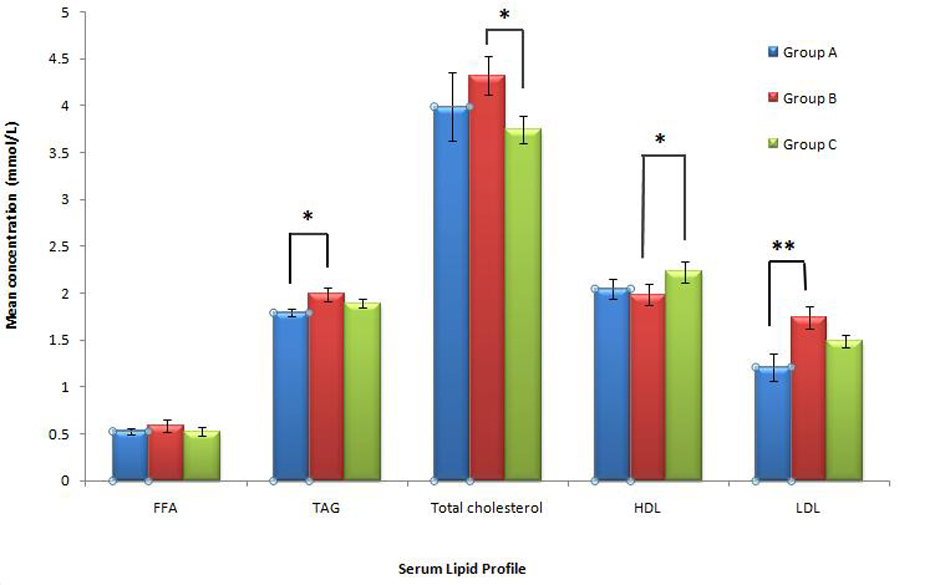 Click for large image | Figure 5. Mean serum free fatty acid (FFA), triglycerides (TAG), total cholesterol, HDL-cholesterol and LDL-cholesterol in rats from Groups A, B and C. (* indicates P< 0.05). Group A, Normal Diet; Group B, High-sucrose diet; Group C, High-sucrose diet + GA. |
Mean serum triglycerides (TAG) level in Group A was found to be lowest, 1.79 ± 0.04 mmol/L. Group B with a value of 1.99 ± 0.07 mmol/L was significantly higher than Group A, representing a 10.9% increase (P = 0.035, P < 0.05). When Group C with a value of 1.89 ± 0.05 mmol/L was compared to Group A, only an insignificant increase of 5.68% was observed (P = 0.116, P > 0.05). Group C also had a lower TAG value of 4.74% compared to Group B (P = 0.301,P > 0.05).
Mean serum total cholesterol concentration (mmol/L) was found to be highest in Group B (4.32 ± 0.21 mmol/L) followed by Group A (3.99 ± 0.38 mmol/L)and the lowest in Group C, (3.75 ± 0.14 mmol/L). With high-sucrose feeding, an insignificant increase was observed in Group B (8.29%) compared to Group A (P = 0.449, P > 0.05). Interestingly, total cholesterol of rats in Group C was reduced by 6.02% compared to Group A (P = 0.536, P > 0.05) and 13.23% lower than Group B (P = 0.034, P < 0.05).
The mean serum high-density lipoprotein (HDL)cholesterol of Groups A, B and C were 2.05 ± 0.10 mmol/L, 1.98 ± 0.11 mmol/L and 2.23 ± 0.11 mmol/L respectively. A non-significant decrease (3.10%) was seen in Group B was compared to Group A (P = 0.291, P > 0.05). However, Group C showed asignificant increase in HDL-cholesterol (12.14% ) compared to Group B (P = 0.042, P < 0.05) while the increase was insignificant (8.67%) compared to Group A (P = 0.244, P > 0.05).
The mean serum low-density lipoprotein (LDL) cholesterol of Groups A, B and C was 1.21 ± 0.14 mmol/L, 1.74 ± 0.12 mmol/L and 1.49 ± 0.07 mmol/L respectively. Likewise, the highest increase of 43.81% was observed in Group B (P = 0.01, P ≤ 0.01) followed by a moderate insignificant increase of 23.1% in Group C compared to Group A (P = 0.10, P > 0.05). The difference of 14.32% between Groups B and C was insignificant (P = 0.088, P > 0.05).
Increased LPL expression in all tissues with GA treatment
In comparing Group A (calibrator) and Group B (target), LPL expression was insignificantly up-regulated in the liver (1.12 ± 0.45 fold), QF (1.24 ± 0.94 fold), AM (1.21 ± 0.70 fold) and VAT (1.23 ± 0.71) (P > 0.05). However, a non-significant down-regulation was observed in kidney (-1.30 ± 0.23 fold), heart (1.32± 0.41 fold) and SAT (-1.39 ± 0.62 fold) (P > 0.05) (Fig. 6).
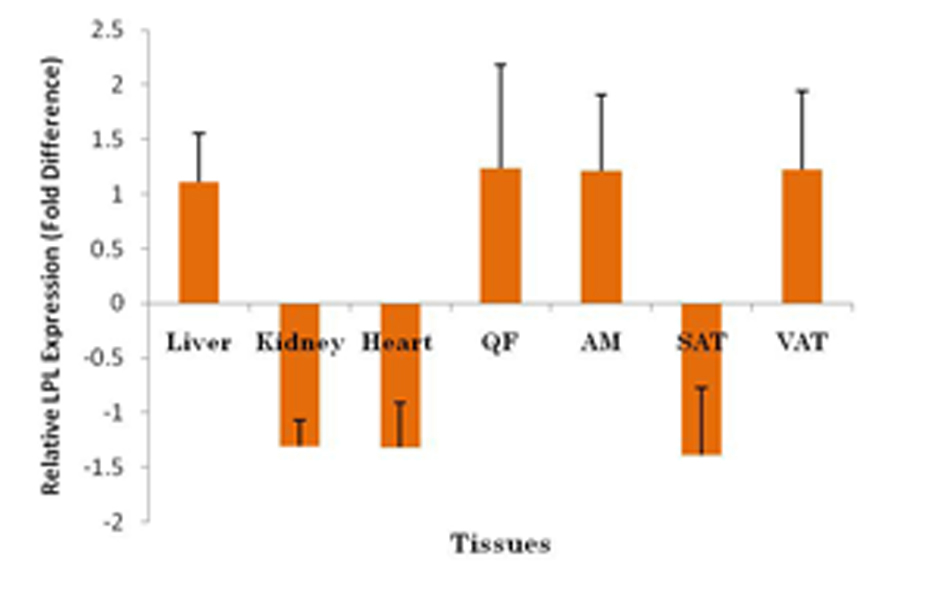 Click for large image | Figure 6. Fold difference of LPL expression in different tissues using BAC as the endogenous reference, tissues from rats fed on normal diet (Group A) as calibrator and tissues from rats fed on high-sucrose diet given GA (Group B) as target. LPL was upregulated in liver, QF, AM and VAT and downregulated in kidney, heart and SAT (P > 0.05). QF, quadriceps femoris; AM, abdominal muscle; SAT, subcutaneous adipose tissue; VAT, visceral adipose tissue; Group A, Normal Diet; Group B, High-sucrose diet; Group C, High-sucrose diet + GA. |
In contrast, comparison between Group B (calibrator) and Group C (target) revealed that LPL expression was upregulated in all tissues with significant increase in the liver. Liver demonstrated the highest increase in fold difference upon GA treatment (2.00 ± 0.34 fold). This was followed by increase in AM (1.24 ± 0.66 fold), SAT (1.18 ± 0.63 fold), kidney (1.16 ± 0.46 fold), VAT (1.08 ± 0.52 fold), QF (1.02 ± 0.82 fold) and heart (1.01 ± 0.47 fold). However, the increases were not significant in all these tissues (P > 0.05) except for the liver (P < 0.05) (Fig. 7).
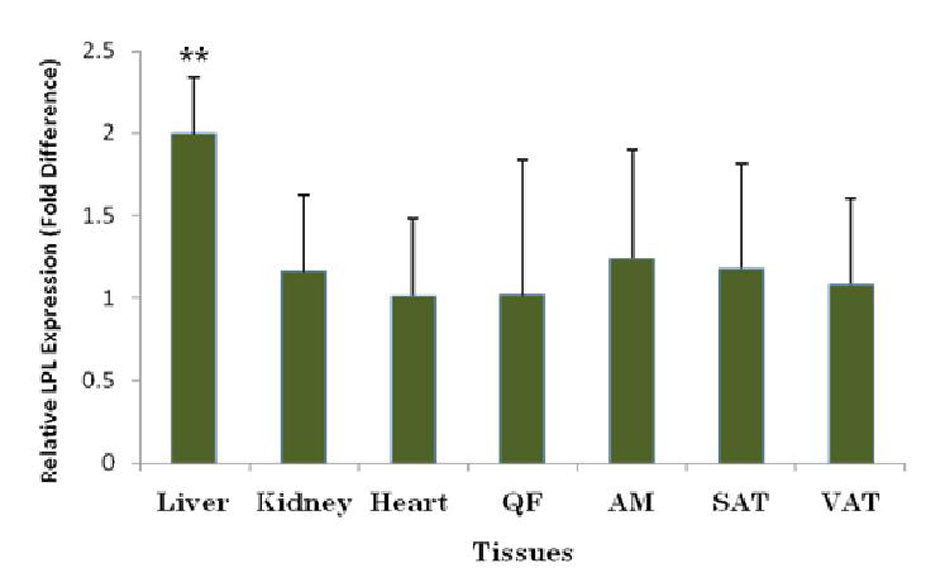 Click for large image | Figure 7. Fold difference of LPL expression in different tissues with BAC as the endogenous reference, tissues from GA-administered high-sucrose-fed group (Group C) as target and tissues from high-sucrose-fed group (Group B) as calibrator. LPL was upregulated in all tissues with significant upregulation in liver. ** indicates P < 0.01. QF, quadriceps femoris; AM, abdominal muscle; SAT, subcutaneous adipose tissue; VAT, visceral adipose tissue; Group A, Normal Diet; Group B, High-sucrose diet; Group C, High-sucrose diet + GA. |
Hexose-6-Phosphate dehydrogenase (H6PDH) activities were lower in all tissues with GA treatment
The mean hepatic H6PDH activities in Groups A, B and C were 1.06 ± 0.11, 1.84 ± 0.12 and 1.21 ± 0.12 units while in the kidney, the values were 6.14 ±0.68, 10.00 ± 1.00 and 6.58 ± 0.98 units respectively. While the SAT of the three groups demonstrated were 610.88 ± 2.38, 19.66 ± 1.87 and 12.54 ± 1.47 units, the VAT recorded values of 16.97 ± 1.01, 23.83 ± 2.04 and 15.59 ± 1.62 units respectively. In the abdominal muscle (AM), the mean H6PDH activitieswere 4.14 ± 0.58, 6.66 ± 0.42 and 4.90 ± 0.50 units respectively for the three groups while in the quadriceps femoris (QF) muscle, the values were 5.44 ±0.65, 10.22 ± 0.86 and 6.27 ± 1.14 units respectively (Fig. 8)
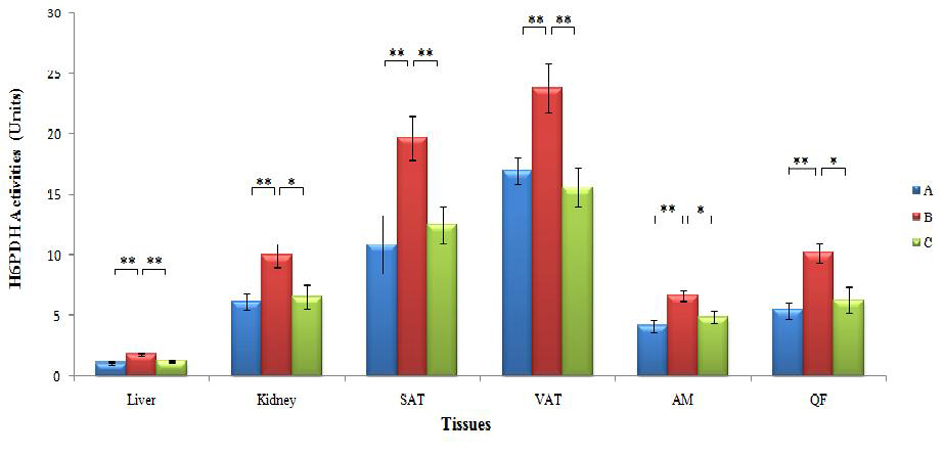 Click for large image | Figure 8. Mean H6PDH activities in the liver, kidneys, SAT, VAT, AM and QF in the three groups of rats (* and ** indicate P < 0.05 and P < 0.01 respectively). H6PDH: hexose-6-phosphate dehydrogenase, SAT: subcutaneous adipose tissue, VAT: visceral adipose tissue, AM: abdominal muscles, QM: quadriceps femoris muscles; Group A, Normal Diet; Group B, High-sucrose diet; Group C, High-sucrose diet + GA. |
When Group B was compared to Group A, the mean H6PDH activities were significantly higher in the liver (73.6%) (P = 0.000, P < 0.01), kidney (62.9%) (P = 0.003, P < 0.01), SAT (80.7%) (P = 0.009, P < 0.01), VAT (40.4%) (P = 0.009, P < 0.01), AM (60.9%) (P = 0.003, P < 0.01) and QF (87.9%) (P = 0.001, P < 0.01). Likewise, the H6PDH activities in Group B were also significantly higher than Group C, 52.1% (P = 0.001, P < 0.01) in the liver, 52.0% (P = 0.024, P < 0.05) in the kidney, 56.8% (P = 0. 008, P < 0.01) in the SAT, 52.9% (P = 0. 006, P < 0.01) in the VAT, 35.9% (P = 0.017, P < 0.05) in the AM and 63.0% (P = 0.015, P < 0.05) in the QF. However, no significant differences were seen between Groups A and C in all the tissues (P > 0.05).
Phosphoenolpyruvate carboxykinase (PEPCK) activities were lower in liver and kidney and higher in SAT and VAT with GA administration
The mean hepatic PEPCK activities in Groups A, B and C were 7.07 ± 0.69, 10.97 ± 0.49 and 8.12 ± 0.63 units respectively while in the kidney, the values were 9.42 ± 1.02, 14.84 ± 1.76 and 10.08 ± 0.86 units respectively (Fig. 9). Group B was significantly higher than Group A in the liver (55.2%) (P = 0.000, P < 0.01) and in the kidney (57.5%) (P = 0.016, P < 0.05). Group B also had a significantly higher PEPCK activity in liver (35.1% ) (P = 0.003, P < 0.01) and kidney (47.2% ) (P = 0.028, P < 0.05) compared to Group C. In both tissues, there was no significant differences between PEPCK activities of Groups A and Group C (P > 0.05).
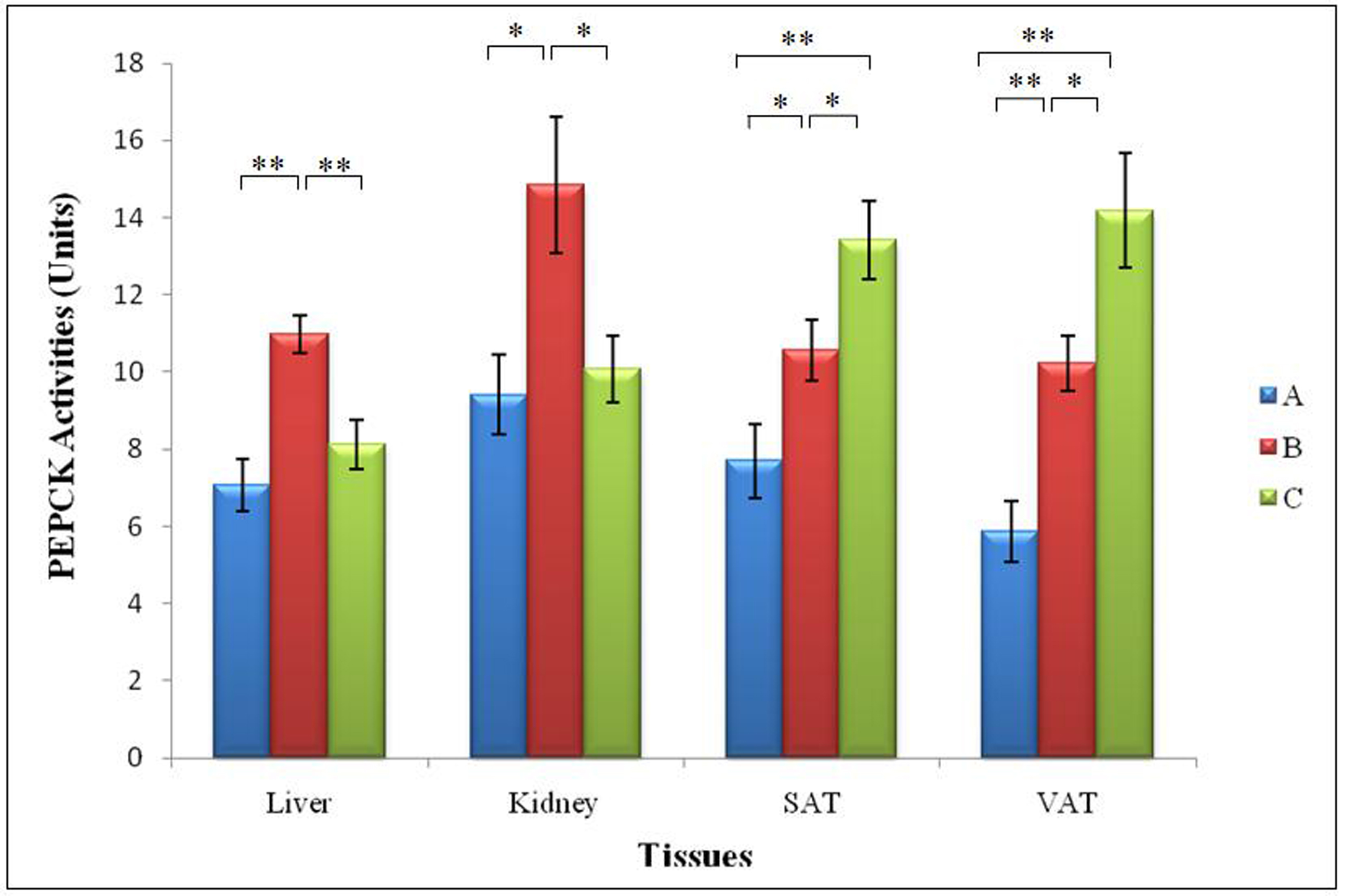 Click for large image | Figure 9. Mean PEPCK activities in the liver, kidneys, SAT and VAT in the three groups of rats. (* and ** indicate P < 0.05 and P < 0.01 respectively). PEPCK: phosphoenolpyruvate carboxykinase, SAT: subcutaneous adipose tissue, VAT: visceral adipose tissue;Group A, Normal Diet; Group B, High-sucrose diet; Group C, High-sucrose diet + GA. |
In the SAT, the mean PEPCK activities of Groups A, B and C were 7.70 ± 0.96, 10.55 ± 0.79 and 13.43 ± 1.00 units respectively while in VAT it was 5.86 ±0.78, 10.22 ± 0.72 and 14.18 ± 1.48 units respectively. Similarly, Group B recorded a higher PEPCK activity in SAT (37.0%) (P = 0.038, P < 0.05) and VAT (74.4%) (P = 0.001, P < 0.01) compared to Group A. Interestingly, PEPCK activity of Group C was significantly higher than in SAT (27.3% ) (P = 0.043, P < 0.05) and VAT ( 38.7% )(P = 0.03, P < 0.05) compared to Group B. Similarly, the difference between Groups C and A was also significant in both SAT (74.4%) (P = 0.001, P < 0.01) and VAT (141%) (P = 0.000, P < 0.01) (Fig. 9).
Glucose-6-Phosphase (G6Pase) activities were toned down with GA treatment
In the liver, the mean hepatic G6Pase activities in Groups A, B and C were 210.12 ± 13.56, 456.04 ± 24.63 and 310.33 ± 32.47 units respectively. Group B had a significant increase (117.0%) compared to Group A (P = 0.000, P < 0.01). The mean hepatic G6Pase activities in Group B was approximately 47.0% higher than that in Group C (P = 0.003, P < 0.01). There was a significant increase (47.7%) in Group C compared to Group A (P = 0.018, P < 0.05) (Fig. 10).
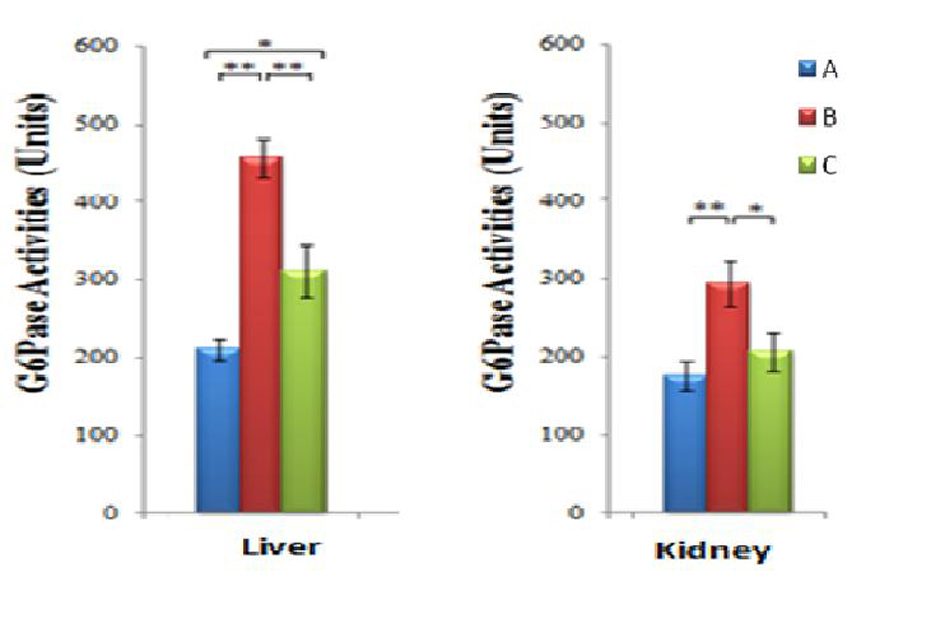 Click for large image | Figure 10. Mean G6Pase activities in kidney and liver of the three groups of rats (* and ** indicate P ≤ 0.05 and P ≤ 0.01 respectively). G6Pase: glucose-6-phosphatase; Group A, Normal Diet; Group B, High-sucrose diet; Group C, High-sucrose diet + GA. |
In the kidneys, the mean G6Pase activities in Groups A, B and C were 175.60 ± 19.46, 293.64 ± 29.36 and 206.59 ± 25.68 units respectively. Group B had a significant increase (67.2%) compared to Group A (P = 0.008, P < 0.01). The mean renal G6Pase activities in Group B was 42.1% higher than that in Group C (P = 0.038, P < 0.05). There was no significant difference between Groups A and C (P = 0.382, P > 0.05) (Fig. 10).
Lower 11β-Hydroxysteroid dehydrogenase (11β-HSD 1) activities in high-sucrose fed group with GA treatment
The mean 11β-HSD 1 activities were highest in the liver tissues over the 4-weeks treatment period in all three groups, with the values of 107.88 ± 8.03, 219.08 ± 19.72 and 119.47 ± 21.071units and in kidney the values were 26.58 ± 1.47, 41.80 ± 1.59 and 32.58 ± 2.06 units respectively. On the other hand, the activities in subcutaneous adipose tissues (SAT) were observed to be 18.14 ± 1.57, 55.75 ± 4.98 and 42.27 ± 3.96 units while in the visceral adipose tissues (VAT), the values were 21.22 ± 2.43, 149.56 ± 28.10 and 53.92 ± 5.01 units respectively. The abdominal muscle tissues (AM) demonstratedvalues of 19.83 ± 2.20, 28.70 ± 3.24 and 12.81 ± 1.67 U respectively whereas the quadriceps femoris (QF) tissues were 12.87 ± 1.74, 17.19 ± 3.15 and 8.65 ± 1.05U respectively for the three groups (Fig. 11).
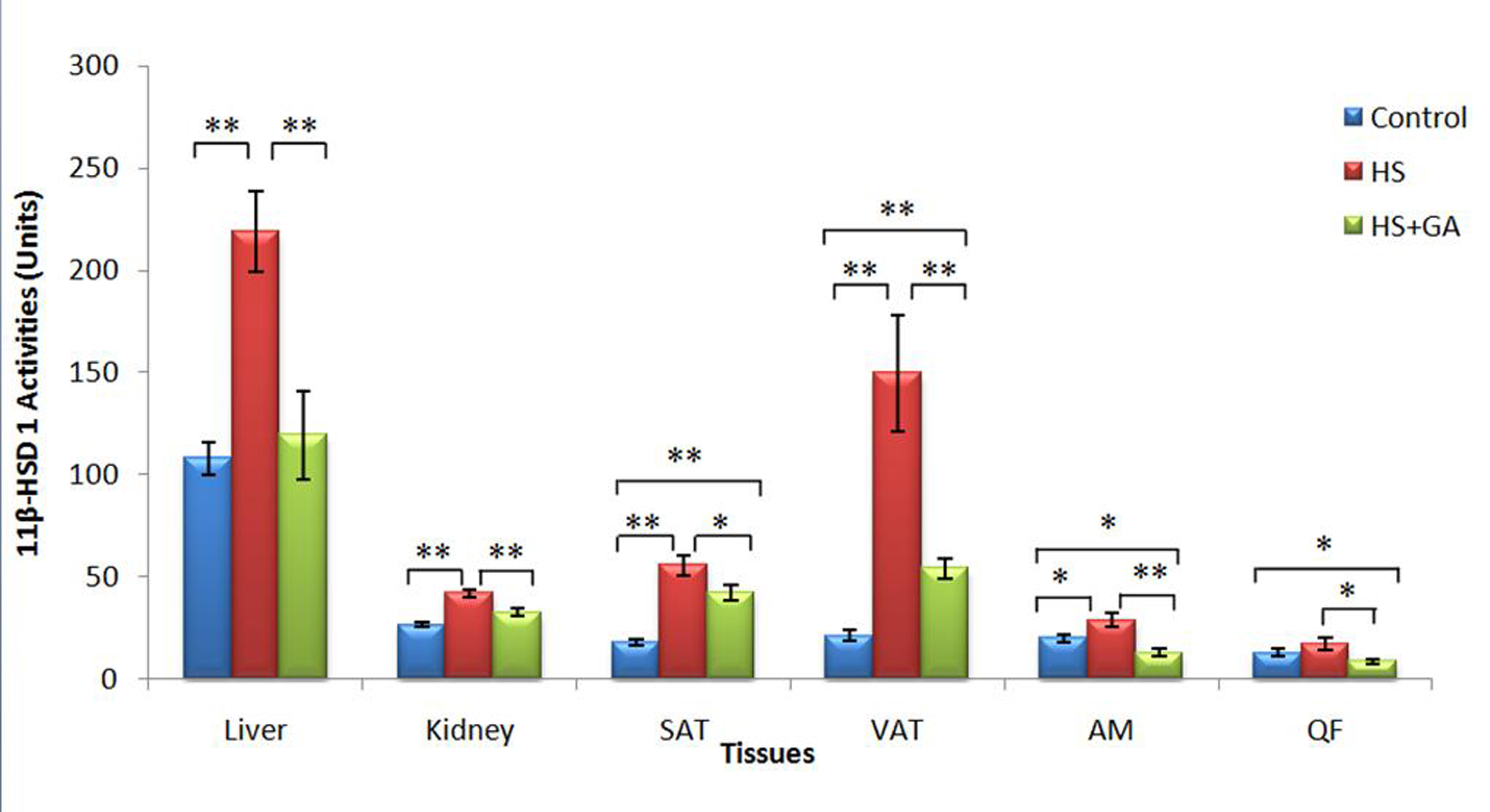 Click for large image | Figure 11. Summary of enzyme activities of 11β-HSD 1 in all tissues of the three experimental groups of rats over the 4-weeks treatment period. (* indicates P ≤ 0.05, ** indicates P ≤ 0.01 respectively).Group A, Normal Diet; Group B, High-sucrose diet; Group C, High-sucrose diet + GA. |
When Group B was compared to Group A, the mean 11β-HSD 1 activities was significantly increased in the liver (103.08%) (P = 0.000; P ≤ 0.01), kidney (57.23%) (P = 0.000; P ≤ 0.01), SAT 207.31% (P = 0.00; P ≤ 0.01), VAT (604.95%) (P = 0.002; P ≤ 0.01, AM (44.72%) (P = 0.039; P ≤ 0.05) and QF (49.69%) (P = 0.05; P ≤ 0.05). All comparisons between Groups B and C were also significant, with significant increases in liver (45.47%) (P = 0.000; P ≤ 0.01), kidney (22.04%) (P = 0.002; P ≤ 0.01), SAT (132.99%) (P = 0.00; P ≤ 0.01), VAT (63.95%) (P = 0.009; P ≤ 0.01), AM (55.36%) (P = 0.001; P ≤ 0.01) and QF (32.78%) (P = 0.031; P ≤ 0.05).
However, when Group A was compared to Group C, a moderate increase was observed in the liver (10.74%) (P = 0.623; P ≤ 0.05and kidney (22.57%) (P = 0.057; P > 0.05), a significant increase was seen in SAT (24.18%) (P = 0.049; P ≤ 0.05) and VAT (63.95%) (P = 0.009; P ≤ 0.01) while a significant decrease was observed in AM (35.40%) (P = 0.018; P ≤ 0.05) and QF (33.62%) (P = 0.206; P > 0.05).
| Discussion | ▴Top |
It has been widely accepted that high-sucrose diets lead to undesirable metabolic abnormalities such as hypertension, hyperglycaemia, insulin resistance and dyslipidaemia. Several studies have utilized animal models such as rabbits [38] mini-pigs [39], mice [40] and rats [41] fed on high-sucrose diet to examine mechanisms underlying diabetes-accelerated dyslipidaemia and atherosclerosis. It has been established that overfeeding of animals with more than 50% of calories from sucrose for approximately four weeks is sufficient to initiate moderate obesity and often results in dyslipidaemia and T2DM [42]. In the present study, rats were fed a high calorie diet with high (60%) calories from sucrose obtained from cane sugar and low amount of calories from fat and protein for four weeks. These rats on high-sucrose diet consumed significantly more calories “per day” compared to the controls which were fed on standard rat chow.
The increased blood pressure observed in high-sucrose fed subjects may be attributed towards the association between hyperinsulinaemic-state and sympathetic nervous system, accumulation of intracellular glyceraldehyde and dihydroxyacetone phosphate and increased level of angiotensin converting enzyme (ACE) [43]. GA administration has also been implicated with mineralocorticoid-like effects characterized by sodium retention, hypokalaemia, hypertension and suppression of renin-angiotensin system and low aldosterone level. This is due to non-selective nature of GA which inhibits both 11β-HSD1 and 11β-HSD2 [44]. However, in our present study, there was no elevation in the blood pressure in Group C with GA administration. This suggests that the increase in blood pressure in rats from Group C is due to the high-sucrose intake and therefore GA administered at 100 mg/kg for 28 days did not contribute towards this increase.
The positive shift in lipid parameters following GA treatment in sucrose-induced-obese rats was similar to that previously reported by Lim et al [34] in lean rats and Eu et al [45] in high-fat-induced obese rats. The hypertriglyceridaemia observed in patients with the MetS and T2DM originates from (i) lipolysis of TAG store from adipose tissue that causes elevated FFA flux to the liver and hence, increased hepatic TAG synthesis and (ii) inhibition of lipolysis of chylomicrons and VLDL due to decreased LPL levels [8]. Our present study indicated that GA treatment has effectively prevented such development through induction of LPL expression in all tissues which promotes catabolism of circulating TAG-rich lipoproteins. More importantly, GA induced a significant increase in HDL levels in the sucrose-induced obese rats. Esterification of free cholesterol is mediated by Lecithin-cholesterol acyltransferase (LCAT) which is bound to HDL-cholesterol. This key antiatherogenic mechanism facilitates removal of excess free cholesterol from the cells in the peripheral tissues and is returnedto the liver and excreted in the bile [46]. Hence, various pharmacological interventions have been focused on raising HDL-cholesterol levels [47]. Thus, the increase in HDL concentration could possibly be a more promising avenue to hinder lower coronary heart disease (CHD) instead of lowering of LDL cholesterol [48].
Fructose, the monosaccharide component of sucrose is highly lipogenic. Fructose provides large amount of hepatic triose-phosphate as precursors for fatty acid synthesis. It has indeed been observed in several studies that hepatic de novo synthesis is stimulated after acute fructose ingestion, with fructose contributing to the synthesis of both the glycerol- and the fatty-acyl parts of VLDL-triglycerides [49]. Fructose may, in addition, increase the expression of key lipogenic enzymes in the liver [50].
Reduced LPL activities in the kidney, heart and SAT were observed in high-sucrose fed subjects. In agreement with our present study, LPL activities in muscle and adipose tissues were seen to be reduced with the onset of IR [6]. The absence of insulin-mediated suppression of lipolysis in adipocytes by hormone sensitive lipase (HSL) promotes the release of fatty acids which in turn inhibit LPL activity [51]. Fatty acids, when in excess, are postulated to bind to LPL and displace it from its binding sites, thereby, rendering them non-functional [52]. Therefore, responses of both LPL and hormone-sensitive-lipase (HSL) are blunted [51].
Fructose may also modulate intracellular lipid deposition known as “ectopic lipids,” i.e., deposition of TAG in the cytoplasm of non-adipose cells, such as hepatocytes and muscle fibers. In rodents, a high-sucrose diet rapidly increased intra-hepatic fat deposition within one week and intra-myocellular lipids on prolonged feeding [53]. This effect of fructose may involve stimulation of de novo lipogenesis through an enhanced intra-hepatic synthesis of triosephosphate precursors, an increased expression of lipogenic genes and LPL in the liver and muscle tissues [54]. Overfeeding promotes increasedmalonyl-CoA which in turn serves as an immediate precursor to promote de novo fatty acid synthesis. Malonyl-CoA also acts as an allosteric inhibitor of the rate-limiting enzyme, carnitine palmitoyltransferase-1 (CPT-1) in the transport of long chain acyl-CoAs (fatty acids) into the mitochondria for β-oxidation. As a result, they are diverted away from mitochondrial oxidation towards biosynthetic enzymes such as glycerol phosphate acyl transferase (GPAT1), diacylglycerol acyl transferase (DGAT1) and serine palmitoyltransferase (SPT1) which promote fatty acids re-esterification into TAGs [55].
Inhibition of 11ß-HSD1 by GA caused a reduction in active glucocorticoids and PPAR agonism properties of GA could be the reasons for the increased LPL expression in all tissues following GA administration. PPARα once activated, leads to a direct up-regulation of LPL expression and also down-regulate apolipoprotein C-III (apo- C-III ), a protein which inhibits TAG hydrolysis by LPL. This results in increased fatty acid uptake and thus reduced serum TAG level as shown by the results from our current study [56, 57]. Elevation of PPARα was observed in 11ß-HSD1 knockout mice by Morton et al [58]. PPARα is physiologically induced by glucocorticoids and its elevation following 11ß-HSD1 inhibition may have arisen from increased circulating plasma glucocorticoids due to impaired negative feedback from the hypothalamic-pituitary-adrenal axis [58]. Therefore, GA-mediated direct or indirect activation of PPAR in concert with inhibition of 11ß-HSD1 may have contributed towards the upregulation of LPL expression in all tissues.
Contrary to fructose-mediated increase in LPL expression which led to tissue lipid accumulation, this GA-mediated increase in LPL expression in all tissues neither lead towards ectopic lipid depositions nor insulin resistance. In addition, the significant upregulation of LPL activities in the liver is probably becausethe liver is centrally involved in metabolism. This may have the highest distribution of GA. Studies on distribution of intravenous (IV) administered GA on rats have confirmed that GA distribution is highest in the liver [59] thus further supporting this postulation. Similarly, LPL expression in the liver was significantly higher in GA-administered rats fed on high-fat diet as well [45].
In our present study, the mean blood glucose concentration was markedly raised with sucrose feeding. This was in accordance with studies on prolonged feeding on high-sucrose diet which increased glucose and insulin responses to a sucrose load [60], increased fasting glycaemia, thereby leading to hepatic insulin resistance in healthy men [61]. Insulin resistance is closely linked to lipid metabolism disorders. Elevated circulating FFA level and high ectopic lipid deposition in IR subjects lead to increased toxic lipid-derived metabolites, such as diacylglycerol, fatty acyl CoA and ceramides. The presence of these metabolites in the intracellular environment leads to higher serine/threonine phosphorylation of insulin receptor substrate-1 (IRS-1), which has been shown to reduce insulin signaling by decreasing GLUT4 translocation to plasma membrane. This in turn reduces insulin-stimulated glucose uptake in peripheral tissues [62].
However, this increase was moderated down in GA-treated rats in Group C. The lower fasting blood glucose may be due to the increased glucose uptake into the adipose tissues and muscles by transporter GLUT4. Saltiel and Kahn [63] reported that insulin sensitizing PPARγ agonist increased the expression of Cb1 associating protein (CAP) which mediates downstream signalling molecules in activating GLUT4 protein mainly in the adipose tissue due to its high expression in this tissue. The enhanced GLUT4 activities in muscles were found to be predominantly mediated by 11β-HSD-1. According to Vegiopoulos and Herzig [64] GC-induced IR in muscles resides in the suppression of glucose uptake mainly through inhibiting translocation of GLUT4 to the cell surface. Studies on 11β-HSD-1 gene knock-out mice also suggest that 11β-HSD-1 inhibition could decrease blood glucose concentrations without risk of hypoglycaemia. Besides increasing insulin-stimulated glucose uptake in tissues, blood glucose concentration was greatly reduced due to the decrease in hepatic glucose production. In T2DM as much as 90% of the hepatic glucose output can be due to accelerated gluconeogenesis. Both the inhibition of 11β-HSD-1 and activation of PPARγ have been demonstrated to decrease expression of gluconeogenic enzymes [32]. Both actions promote gene expressions of fatty acid binding protein (FABP), LPL, acyl-CoA synthase (ACoAS) and PEPCK. This will lead to increased re-esterification of FFA and enhance fat storage in the adipose tissues. Both 11β-HSD1 inhibition and PPARγ activation also suppress the genes that induce lipolysis and the release of FFAs, such as β3-adrenergic receptor, leptin and TNF-α. This causes reduced circulating FFA levels and amelioration of FFA-induced hepatic and peripheral IR, resulting in increased hepatic and peripheral insulin sensitivity and improved glucose disposal [65].
Results from our present study have revealed that high-sucrose diet resulted in higher H6PDH activities in all six tissues. This result is in agreement withLondon et al [66] who observed increased H6PDH mRNA levels in the liver and adipose tissues of Sprague-Dawley rats. High-sucrose increases the substrate, glucose-6-phosphate (G6P) and the availability for H6PDH, thereby leading to increased production of cofactor NADPH via the pentose phosphate pathway. The increased NADPH subsequently promotes the reductase activity of 11β-HSD1, resulting in elevated production of active GCs [66]. This postulation is further confirmed by elevated 11β-HSD1 activity observed in all tissues. Moreover, fructose enhances glucose phosphorylation in the liver. Fructose, derived from high-sucrose diet, is converted to fructose-1-phosphate which then binds to glucose kinase regulatory protein (GKRP) and causes the dissociation of glucokinase (GK) from the glucokinase binding protein (GKBP). GK is translocated from the nucleus to the cytoplasm, resulting in an increase in the conversion of glucose to G6P [67]. This increases the substrate availability for H6PDH activities [68]. Lower H6PDH activities observed in ratswith GA administration is probably due to the inhibitory effect of GA on 11β-HSD1. This leads to a reduction in the generation of cofactor NADP+ therebydecreasing the H6PDH activities [69]. GA administration leads to reduced PEPCK and G6Pase expression, which subsequently decrease the rate of hepatic gluconeogenesis. GA’s activation of PPARγ also improves hepatic and peripheral insulin sensitivity and glucose uptake. Both effects of GA lead to reduced hepatic glycogen store and circulating blood glucose, leading to a decrease in the production of G6P from glucose and glycogen through glycolysis and glycogenolysis. As a consequence, the availability of G6P for H6PDH decreases, contributing to a reduction in H6PDH activities [69].
As aforementioned, PEPCK is a key regulatory enzyme involved in two metabolic processes: gluconeogenesis, which occurs in the liver and kidney, and glyceroneogenesis, which occurs in the adipose tissues [70]. Our present study shows elevated PEPCK activities in all tissues with sucrose feeding. Increased expression of hepatic PEPCK gene was also observed in diabetic mice, characterized by elevated hepatic glucose output [71]. This could be due to increased production of GCs which stimulates the hepatic and renal PEPCK activities by promoting the assembly of several transcription factors (TFs),hepatocyte nuclear factors (HNF-3β and HNF-4α), Ccaat-enhancer-binding proteins (C/EBP β) and Forkhead box protein O1 (FOXO1), on the PEPCK gene promoter [72]. However, Cassuto et al found that PEPCK activities are only stimulated by GCs in the liver and kidneys, but are suppressed in adipose tissues [73]. Our present results is contrary to the above, where significantly increased PEPCK activities were observed in all tissues of Group B. Increased PEPCK activities in SAT and VAT in group B could be attributed to the FFA-induced activation of PPARγ. PPARγ is predominantly expressed in adipose tissues.During high-sucrose feeding, elevated GC-induced lipolysis causes activation of re-esterification to prevent an all-out release of FFA into the bloodstream. As a consequence, glyceroneogenesis is activated to produce G3P, which is required for fatty acid re-esterification. Under this condition, elevated circulating FFA would activate PPARγ, which binds as a heterodimer complex with RXRα to the PPAR response element (PPRE) on the PEPCK gene promoter. This results in increased PEPCK gene transcription and a corresponding increase in its activities in the adipose tissues [74].
Group C had increased hepatic and renal PEPCK activities compared to group A, but the elevations were smaller compared to group B. This indicated the ability of GA to improve hyperglycaemia through GC-stimulated PEPCK activities in gluconeogenesis. GA administration leads to reduced production of active GCs. This results in a downregulation in the PEPCK gene transcription, thereby decreasing PEPCK activities and gluconeogenesis rate [32]. In addition, activation of PPARγ was also found to downregulate the gene expression of PEPCK in the liver and kidneys, resulting in decreased gluconeogenesis [75]. GA, which is a triterpenoid saponin, has been found to be an agonist of PPARγ [76]. Therefore, GA’s activation of PPARγ would lead to downregulation of PEPCK gene transcription and this would also account for the reduced PEPCK activities observed in GA-treated rats compared to non-GA-treated rats.
G6Pase is mainly expressed in gluconeogenic tissues such as the liver and kidneys. It is a membrane-bound enzyme with a multi-component system consisting of G6PC and G6PT which are located in the ER [77]. Similar to H6PDH, G6Pase requires G6P as substrate. It catalyses the final step of gluconeogenesis and glycogenolysis, which is the hydrolysis of G6P to glucose [78]. Following high-sucrose feeding, an increase in 6-carbon sugars (glucose and fructose) would increase the availability of substrate G6P for G6Pase. This would lead to increased G6Pase activities and elevated glucose output [79]. In addition, increased production of active GCs during high-sucrose feeding has been found to induce the gene transcription of G6Pase. This involves the binding of GR and cofactors to G6Pase gene promoter and increased co-activation of PGC-1α with GR, FOXO1 and HNF-4, which promote the G6Pase gene transcription [80]. High circulating FFA has also been associated with increased G6Pase mRNA and activities in diabetic condition. Elevated circulating FFA exerts a PPAR-dependent allosteric stimulation on G6Pase, resulting in increased G6Pase expression and gluconeogenesis [81]. Smaller elevation in G6Pase activities were observed in GA-treated rats compared to non-treated rats on high-sucrose diet. Besides the reduction in active GCs, another possible mechanism which might account for reduced G6Pase activities in the liver and kidneys could be the actions of PPARα in improving glucose and lipid metabolism and enhancing whole body insulin sensitivity. Treatment with PPARα agonist was found to significantly reduce the mRNA level of G6Pase, leading to decreased gluconeogenesis. This suggests the possible role of GA as a PPARα agonist in the modulation of G6Pase activities [65].
Despite significantly higher serum insulin level in sucrose-fed rats, insulin failed to suppress the PEPCK and G6Pase activities in all the studied tissues. This could be due to reduced responsiveness of the tissues to insulin action due to increased circulating FFA levels. This will impair the insulin-stimulated suppression of PEPCK and G6Pase activities [81].
Conclusion
Our study has indicated that high-sucrose feeding could induce hypertension, hyperglycaemia and insulin resistance. GA administration at a dosage of 100mg/kg for 28 days could ameliorate these symptoms by regulating the glucose metabolism and causing an apparent hypotriglyceridaemic and HDL-raising effect. Upregulation of LPL expression in all tissues may promote proper tissue partitioning of fat and retards the development of CVD and subsequently MetS. Moreover, selective induction of PEPCK activities and moderated increase in H6PDH and G6Pase activites accompanied by improvement in insulin sensitivity were achieved with GA administration without any hypertensive effect. This suggests that GA could be considered as both an anti-hyperglycaemic and anti-dyslipidaemic compound.
Conflict of Interest
There is no potential conflict of interest relevant to this article.
| References | ▴Top |
- Janus E. (2004) Metabolic syndrome and its relevance to Asia. International Congress Series 1262: 535-537.
- Balkau B, Valensi P, Eschwege E, Slama G. A review of the metabolic syndrome. Diabetes Metab. 2007;33(6):405-413.
pubmed doi - Grundy SM. Metabolic syndrome pandemic. Arterioscler Thromb Vasc Biol. 2008;28(4):629-636.
pubmed doi - Aguilera AA, Diaz GH, Barcelata ML, Guerrero OA, Ros RM. Effects of fish oil on hypertension, plasma lipids, and tumor necrosis factor-alpha in rats with sucrose-induced metabolic syndrome. J Nutr Biochem. 2004;15(6):350-357.
pubmed doi - London E, Castonguay TW. Diet and the role of 11beta-hydroxysteroid dehydrogenase-1 on obesity. J Nutr Biochem. 2009;20(7):485-493.
pubmed doi - Pollare T, Vessby B, Lithell H. Lipoprotein lipase activity in skeletal muscle is related to insulin sensitivity. Arterioscler Thromb. 1991;11(5):1192-1203.
pubmed doi - Preiss-Landl K, Zimmermann R, Hammerle G, Zechner R. Lipoprotein lipase: the regulation of tissue specific expression and its role in lipid and energy metabolism. Curr Opin Lipidol. 2002;13(5):471-481.
pubmed doi - Lann D, LeRoith D. Insulin resistance as the underlying cause for the metabolic syndrome. Med Clin North Am. 2007;91(6):1063-1077, viii.
pubmed - Weinstock PH, Bisgaier CL, Aalto-Setala K, Radner H, Ramakrishnan R, Levak-Frank S, Essenburg AD, et al. Severe hypertriglyceridemia, reduced high density lipoprotein, and neonatal death in lipoprotein lipase knockout mice. Mild hypertriglyceridemia with impaired very low density lipoprotein clearance in heterozygotes. J Clin Invest. 1995;96(6):2555-2568.
pubmed doi - Zechner R. The tissue-specific expression of lipoprotein lipase: implications for energy and lipoprotein metabolism. Curr Opin Lipidol. 1997;8(2):77-88.
pubmed doi - Ferreira LD, Pulawa LK, Jensen DR, Eckel RH. Overexpressing human lipoprotein lipase in mouse skeletal muscle is associated with insulin resistance. Diabetes. 2001;50(5):1064-1068.
pubmed doi - Reynolds RM, Walker BR, Syddall HE, Andrew R, Wood PJ, Whorwood CB, Phillips DI. Altered control of cortisol secretion in adult men with low birth weight and cardiovascular risk factors. J Clin Endocrinol Metab. 2001;86(1):245-250.
pubmed doi - Wang M. The role of glucocorticoid action in the pathophysiology of the Metabolic Syndrome. Nutr Metab (Lond). 2005;2(1):3.
pubmed doi - Hewitt KN, Walker EA, Stewart PM. Minireview: hexose-6-phosphate dehydrogenase and redox control of 11{beta}-hydroxysteroid dehydrogenase type 1 activity. Endocrinology. 2005;146(6):2539-2543.
pubmed doi - Lavery GG, Hauton D, Hewitt KN, Brice SM, Sherlock M, Walker EA, Stewart PM. Hypoglycemia with enhanced hepatic glycogen synthesis in recombinant mice lacking hexose-6-phosphate dehydrogenase. Endocrinology. 2007;148(12):6100-6106.
pubmed doi - Nashev LG, Chandsawangbhuwana C, Balazs Z, Atanasov AG, Dick B, Frey FJ, Baker ME, et al. Hexose-6-phosphate dehydrogenase modulates 11beta-hydroxysteroid dehydrogenase type 1-dependent metabolism of 7-keto- and 7beta-hydroxy-neurosteroids. PLoS One. 2007;2(6):e561.
pubmed doi - Lambillotte C, Gilon P, Henquin JC. Direct glucocorticoid inhibition of insulin secretion. An in vitro study of dexamethasone effects in mouse islets. J Clin Invest. 1997;99(3):414-423.
pubmed doi - Nelson RA, Bremer AA. Insulin resistance and metabolic syndrome in the pediatric population. Metab Syndr Relat Disord. 2010;8(1):1-14.
pubmed doi - Andrews RC, Walker BR. Glucocorticoids and insulin resistance: old hormones, new targets. Clin Sci (Lond). 1999;96(5):513-523.
pubmed doi - Kolovou GD, Anagnostopoulou KK, Cokkinos DV. Pathophysiology of dyslipidaemia in the metabolic syndrome. Postgrad Med J. 2005;81(956):358-366.
pubmed doi - Masoudi FA, Inzucchi SE, Wang Y, Havranek EP, Foody JM, Krumholz HM. Thiazolidinediones, metformin, and outcomes in older patients with diabetes and heart failure: an observational study. Circulation. 2005;111(5):583-590.
pubmed doi - Bajaj M, Suraamornkul S, Pratipanawatr T, Hardies LJ, Pratipanawatr W, Glass L, Cersosimo E, et al. Pioglitazone reduces hepatic fat content and augments splanchnic glucose uptake in patients with type 2 diabetes. Diabetes. 2003;52(6):1364-1370.
pubmed doi - Krent A.J. and Bailey, C.J. (2006) Oral antidiabetic agents. Current role in type 2 diabetes mellitus. Drugs 65: 385-411.
- Post SM, Duez H, Gervois PP, Staels B, Kuipers F, Princen HM. Fibrates suppress bile acid synthesis via peroxisome proliferator-activated receptor-alpha-mediated downregulation of cholesterol 7alpha-hydroxylase and sterol 27-hydroxylase expression. Arterioscler Thromb Vasc Biol. 2001;21(11):1840-1845.
pubmed doi - Yki-Jarvinen H. Thiazolidinediones. N Engl J Med. 2004;351(11):1106-1118.
pubmed doi - Shimono K, Tsutsumi K, Yaguchi H, Omura M, Sasano H, Nishikawa T. Lipoprotein lipase promoting agent, NO-1886, modulates adrenal functions: species difference in effects of NO-1886 on steroidogenesis. Steroids. 1999;64(7):453-459.
pubmed doi - Isbrucker RA, Burdock GA. Risk and safety assessment on the consumption of Licorice root (Glycyrrhiza sp.), its extract and powder as a food ingredient, with emphasis on the pharmacology and toxicology of glycyrrhizin. Regul Toxicol Pharmacol. 2006;46(3):167-192.
pubmed doi - Ploeger BA, Meulenbelt J, DeJongh J. Physiologically based pharmacokinetic modeling of glycyrrhizic acid, a compound subject to presystemic metabolism and enterohepatic cycling. Toxicol Appl Pharmacol. 2000;162(3):177-188.
pubmed doi - Tomlinson JW, Sherlock M, Hughes B, Hughes SV, Kilvington F, Bartlett W, Courtney R, et al. Inhibition of 11beta-hydroxysteroid dehydrogenase type 1 activity in vivo limits glucocorticoid exposure to human adipose tissue and decreases lipolysis. J Clin Endocrinol Metab. 2007;92(3):857-864.
pubmed doi - Wang Y, Porter WW, Suh N, Honda T, Gribble GW, Leesnitzer LM, Plunket KD, et al. A synthetic triterpenoid, 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO), is a ligand for the peroxisome proliferator-activated receptor gamma. Mol Endocrinol. 2000;14(10):1550-1556.
pubmed doi - Sato M, Tai T, Nunoura Y, Yajima Y, Kawashima S, Tanaka K. Dehydrotrametenolic acid induces preadipocyte differentiation and sensitizes animal models of noninsulin-dependent diabetes mellitus to insulin. Biol Pharm Bull. 2002;25(1):81-86.
pubmed doi - Alberts P, Engblom L, Edling N, Forsgren M, Klingstrom G, Larsson C, Ronquist-Nii Y, et al. Selective inhibition of 11beta-hydroxysteroid dehydrogenase type 1 decreases blood glucose concentrations in hyperglycaemic mice. Diabetologia. 2002;45(11):1528-1532.
pubmed doi - Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18(6):499-502.
pubmed - Lim WY, Chia YY, Liong SY, Ton SH, Kadir KA, Husain SN. Lipoprotein lipase expression, serum lipid and tissue lipid deposition in orally-administered glycyrrhizic acid-treated rats. Lipids Health Dis. 2009;8:31.
pubmed - Petrescu I, Bojan O, Saied M, Barzu O, Schmidt F, Kuhnle HF. Determination of phosphoenolpyruvate carboxykinase activity with deoxyguanosine 5'-diphosphate as nucleotide substrate. Anal Biochem. 1979;96(2):279-281.
pubmed doi - Banhegyi G, Benedetti A, Fulceri R, Senesi S. Cooperativity between 11beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase in the lumen of the endoplasmic reticulum. J Biol Chem. 2004;279(26):27017-27021.
pubmed doi - Csala M, Margittai E, Senesi S, Gamberucci A, Banhegyi G, Mandl J, Benedetti A. Inhibition of hepatic glucose 6-phosphatase system by the green tea flavanol epigallocatechin gallate. FEBS Lett. 2007;581(8):1693-1698.
pubmed doi - Yin W, Yuan Z, Wang Z, Yang B, Yang Y. A diet high in saturated fat and sucrose alters glucoregulation and induces aortic fatty streaks in New Zealand White rabbits. Int J Exp Diabetes Res. 2002;3(3):179-184.
pubmed doi - Xi S, Yin W, Wang Z, Kusunoki M, Lian X, Koike T, Fan J, et al. A minipig model of high-fat/high-sucrose diet-induced diabetes and atherosclerosis. Int J Exp Pathol. 2004;85(4):223-231.
pubmed doi - Shafrir E, Ziv E, Kalman R. Nutritionally induced diabetes in desert rodents as models of type 2 diabetes: Acomys cahirinus (spiny mice) and Psammomys obesus (desert gerbil). ILAR J. 2006;47(3):212-224.
pubmed - Yang M, Wang C, Chen H. Green, oolong and black tea extracts modulate lipid metabolism in hyperlipidemia rats fed high-sucrose diet. J Nutr Biochem. 2001;12(1):14-20.
pubmed doi - Kim JY, Nolte LA, Hansen PA, Han DH, Kawanaka K, Holloszy JO. Insulin resistance of muscle glucose transport in male and female rats fed a high-sucrose diet. Am J Physiol. 1999;276(3 Pt 2):R665-672.
pubmed - Coelho MS, Lopes KL, Freitas Rde A, de Oliveira-Sales EB, Bergasmaschi CT, Campos RR, Casarini DE, et al. High sucrose intake in rats is associated with increased ACE2 and angiotensin-(1-7) levels in the adipose tissue. Regul Pept. 2010;162(1-3):61-67.
pubmed doi - Stormer FC, Reistad R, Alexander J. Glycyrrhizic acid in liquorice—evaluation of health hazard. Food Chem Toxicol. 1993;31(4):303-312.
pubmed doi - Eu CH, Lim WY, Ton SH, bin Abdul Kadir K. Glycyrrhizic acid improved lipoprotein lipase expression, insulin sensitivity, serum lipid and lipid deposition in high-fat diet-induced obese rats. Lipids Health Dis. 2010;9:81.
pubmed - Barter P. (2005) The role of HDL-cholesterol in preventing atherosclerotic disease. European Heart Journal Supplements 7: F4-F8.
- Assmann G, Nofer JR. Atheroprotective effects of high-density lipoproteins. Annu Rev Med. 2003;54:321-341.
pubmed - Dullens SP, Plat J, Mensink RP. Increasing apoA-I production as a target for CHD risk reduction. Nutr Metab Cardiovasc Dis. 2007;17(8):616-628.
pubmed doi - Chong MF, Fielding BA, Frayn KN. Mechanisms for the acute effect of fructose on postprandial lipemia. Am J Clin Nutr. 2007;85(6):1511-1520.
pubmed - Parks EJ, Hellerstein MK. Carbohydrate-induced hypertriacylglycerolemia: historical perspective and review of biological mechanisms. Am J Clin Nutr. 2000;71(2):412-433.
pubmed - Franssen R, Monajemi H, Stroes ES, Kastelein JJ. Obesity and dyslipidemia. Endocrinol Metab Clin North Am. 2008;37(3):623-633, viii.
pubmed - Pulinilkunnil T, Rodrigues B. Cardiac lipoprotein lipase: metabolic basis for diabetic heart disease. Cardiovasc Res. 2006;69(2):329-340.
pubmed doi - Pagliassotti MJ, Prach PA, Koppenhafer TA, Pan DA. Changes in insulin action, triglycerides, and lipid composition during sucrose feeding in rats. Am J Physiol. 1996;271(5 Pt 2):R1319-1326.
pubmed - Kim JK, Fillmore JJ, Chen Y, Yu C, Moore IK, Pypaert M, Lutz EP, et al. Tissue-specific overexpression of lipoprotein lipase causes tissue-specific insulin resistance. Proc Natl Acad Sci U S A. 2001;98(13):7522-7527.
pubmed doi - Muoio DM, Newgard CB. Obesity-related derangements in metabolic regulation. Annu Rev Biochem. 2006;75:367-401.
pubmed - Lee CH, Olson P, Evans RM. Minireview: lipid metabolism, metabolic diseases, and peroxisome proliferator-activated receptors. Endocrinology. 2003;144(6):2201-2207.
pubmed doi - Tenenbaum A, Fisman EZ, Motro M. Metabolic syndrome and type 2 diabetes mellitus: focus on peroxisome proliferator activated receptors (PPAR). Cardiovasc Diabetol. 2003;2:4.
pubmed - Morton NM, Holmes MC, Fievet C, Staels B, Tailleux A, Mullins JJ, Seckl JR. Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in 11beta-hydroxysteroid dehydrogenase type 1 null mice. J Biol Chem. 2001;276(44):41293-41300.
pubmed doi - Zeng CX, Yang Q, Hu Q. A comparison of the distribution of two glycyrrhizic acid epimers in rat tissues. Eur J Drug Metab Pharmacokinet. 2006;31(4):253-258.
pubmed doi - Hallfrisch J, Ellwood KC, Michaelis OEt, Reiser S, O'Dorisio TM, Prather ES. Effects of dietary fructose on plasma glucose and hormone responses in normal and hyperinsulinemic men. J Nutr. 1983;113(9):1819-1826.
pubmed - Faeh D, Minehira K, Schwarz JM, Periasamy R, Park S, Tappy L. Effect of fructose overfeeding and fish oil administration on hepatic de novo lipogenesis and insulin sensitivity in healthy men. Diabetes. 2005;54(7):1907-1913.
pubmed doi - Wassink AM, Olijhoek JK, Visseren FL. The metabolic syndrome: metabolic changes with vascular consequences. Eur J Clin Invest. 2007;37(1):8-17.
pubmed doi - Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799-806.
pubmed doi - Vegiopoulos A, Herzig S. Glucocorticoids, metabolism and metabolic diseases. Mol Cell Endocrinol. 2007;275(1-2):43-61.
pubmed doi - Kim HI, Ahn YH. Role of peroxisome proliferator-activated receptor-gamma in the glucose-sensing apparatus of liver and beta-cells. Diabetes. 2004;53 Suppl 1:S60-65.
pubmed - London E, Lala G, Berger R, Panzenbeck A, Kohli AA, Renner M, Jackson A, et al. Sucrose access differentially modifies 11beta-hydroxysteroid dehydrogenase-1 and hexose-6-phosphate dehydrogenase message in liver and adipose tissue in rats. J Nutr. 2007;137(12):2616-2621.
pubmed - Agius L. The physiological role of glucokinase binding and translocation in hepatocytes. Adv Enzyme Regul. 1998;38:303-331.
pubmed - Senesi S, Legeza B, Balazs Z, Csala M, Marcolongo P, Kereszturi E, Szelenyi P, et al. Contribution of fructose-6-phosphate to glucocorticoid activation in the endoplasmic reticulum: possible implication in the metabolic syndrome. Endocrinology. 2010;151(10):4830-4839.
pubmed doi - Liu Y, Nakagawa Y, Wang Y, Liu L, Du H, Wang W, Ren X, et al. Reduction of hepatic glucocorticoid receptor and hexose-6-phosphate dehydrogenase expression ameliorates diet-induced obesity and insulin resistance in mice. J Mol Endocrinol. 2008;41(2):53-64.
pubmed doi - Agca C, Greenfield RB, Hartwell JR, Donkin SS. Cloning and characterization of bovine cytosolic and mitochondrial PEPCK during transition to lactation. Physiol Genomics. 2002;11(2):53-63.
pubmed - Hanson RW, Reshef L. Regulation of phosphoenolpyruvate carboxykinase (GTP) gene expression. Annu Rev Biochem. 1997;66:581-611.
pubmed - Shao J, Qiao L, Janssen RC, Pagliassotti M, Friedman JE. Chronic hyperglycemia enhances PEPCK gene expression and hepatocellular glucose production via elevated liver activating protein/liver inhibitory protein ratio. Diabetes. 2005;54(4):976-984.
pubmed doi - Cassuto H, Olswang Y, Livoff AF, Nechushtan H, Hanson RW, Reshef L. Involvement of HNF-1 in the regulation of phosphoenolpyruvate carboxykinase gene expression in the kidney. FEBS Lett. 1997;412(3):597-602.
pubmed doi - Sugden MC, Zariwala MG, Holness MJ. PPARs and the orchestration of metabolic fuel selection. Pharmacol Res. 2009;60(3):141-150.
pubmed doi - Way JM, Harrington WW, Brown KK, Gottschalk WK, Sundseth SS, Mansfield TA, Ramachandran RK, et al. Comprehensive messenger ribonucleic acid profiling reveals that peroxisome proliferator-activated receptor gamma activation has coordinate effects on gene expression in multiple insulin-sensitive tissues. Endocrinology. 2001;142(3):1269-1277.
pubmed doi - Yoke Yin C, So Ha T, Abdul Kadir K. Effects of Glycyrrhizic Acid on Peroxisome Proliferator-Activated Receptor Gamma (PPARgamma), Lipoprotein Lipase (LPL), Serum Lipid and HOMA-IR in Rats. PPAR Res. 2010;2010 :530265.
pubmed - Hutton JC, O'Brien RM. Glucose-6-phosphatase catalytic subunit gene family. J Biol Chem. 2009;284(43):29241-29245.
pubmed doi - Csala M, Banhegyi G, Benedetti A. Endoplasmic reticulum: a metabolic compartment. FEBS Lett. 2006;580(9):2160-2165.
pubmed doi - van Schaftingen E, Gerin I. The glucose-6-phosphatase system. Biochem J. 2002;362(Pt 3):513-532.
pubmed doi - Yabaluri N, Bashyam MD. Hormonal regulation of gluconeogenic gene transcription in the liver. J Biosci. 2010;35(3):473-484.
pubmed doi - Lam TK, Carpentier A, Lewis GF, van de Werve G, Fantus IG, Giacca A. Mechanisms of the free fatty acid-induced increase in hepatic glucose production. Am J Physiol Endocrinol Metab. 2003;284(5):E863-873.
pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.