

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website https://www.jofem.org |
Case Report
Volume 12, Number 1, February 2022, pages 40-48

Cushing’s Disease of Unknown Etiology: A Clinical Case Report
Joao Martin Martinsa, e ![]() , Catarina Martins Maiab, Jose Miguensc, Dolores Lopez Presad
, Catarina Martins Maiab, Jose Miguensc, Dolores Lopez Presad
aEndocrine Department, Santa Maria Hospital and Lisbon Medical School, Lisbon, Portugal
bInternal Medicine Department, Jacobi Medical Center and Albert Einstein College of Medicine, Bronx, NY, USA
cNeurosurgery Department, Santa Maria Hospital and Lisbon Medical School, Lisbon, Portugal
dPathology Department, Santa Maria Hospital and Lisbon Medical School, Lisbon, Portugal
eCorresponding Author: Joao Martin Martins, Endocrine Department, Santa Maria Hospital and Lisbon Medical School, Lisbon, Portugal
Manuscript submitted October 21, 2021, accepted December 9, 2021, published online February 15, 2022
Short title: Cushing’s Disease of Unknown Etiology
doi: https://doi.org/10.14740/jem784
| Abstract | ▴Top |
Cushing’s disease (CD) remains a diagnostic and therapeutic challenge. Different subtypes may be recognized that will offer insight in this complex situation. We describe an atypical case that we assume is a common variation albeit with no previous formal description. A young female patient presented with minimal clinical features of CD, but increased adrenocorticotropic hormone (ACTH) and cortisol levels, with maintained circadian rhythm that was not suppressed either after the rapid dexamethasone or the prolonged low-dose dexamethasone tests, but suppressed with the prolonged high-dose dexamethasone test and presented a flat ACTH and cortisol response after the corticotropin-releasing hormone (CRH) test. A diffuse enlarged pituitary gland with thickened pituitary stalk was present and a mixed corticotroph adenoma was removed. CD persisted despite now normal pituitary morphology, except for pituitary stalk widening. Plasma levels of CRH were low and no abnormalities were found in the coding region or flanking introns of glucocorticoid receptor (GCR) gene (NR3C1). Somatostatin receptors were not present in the octreoscan, and treatment with cabergoline or somatostatin analogs was ineffective. Morbidity and mortality are increased in CD even in patients successfully treated and in remission. Despite early success in over 80% of the patients, in the long term CD recurs in almost 50% of the patients. Defining subtypes of CD may help elucidate mechanisms of the disease. We propose a new variant that we assume is common. Furthermore adaptation to chronic hypercortisolism is present.
Keywords: Cushing’s disease variant; Dexamethasone test; Corticotroph adenoma; GCR gene; Subtypes
| Introduction | ▴Top |
Hypercortisolism is a common condition in everyday clinical practice, but most cases are either obvious or not immediately relevant by themselves: hypercortisolism due to the use of supraphysiologic doses of corticosteroids in the treatment of inflammatory and immune-mediated disorders [1], hypercortisolism in relation to the ectopic secretion of adrenocorticotropic hormone (ACTH) in common malignancies like the small cell adenocarcinoma of the lung or more rarely pancreatic neuroendocrine tumors [2, 3], and subclinical hypercortisolism in regard to incidentally found adrenal nodules, although in this case, clinical importance is still a matter of ongoing debate [4].
Hypercortisolism may also be particularly common in selected groups of patients like those with diabetes mellitus (< 10%), high blood pressure (1%), osteoporosis (11%) or in women with irregular menses and hirsutism (1%) [5-9].
Outside those specific settings hypercortisolism is much less common, with two to three new cases per million per year [10]. Diagnostic approach always includes discriminating between ACTH-dependent (around 70% of all cases) and ACTH-independent Cushing’s syndrome (CS) [11, 12], even if there is still a significant delay in diagnosis [13] and an increased morbidity and mortality before and even after diagnosis and treatment [14].
Cushing’s disease (CD) with an estimated incidence of around 1.5 per million per year, female preponderance (3:1) and peaking at ages between 25 and 45 years accounts for most of the cases of ACTH-dependent CS [10, 15, 16]. CD is due in most cases to a monoclonal pituitary microadenoma, sometimes with oncogenic triggering events in the cyclin or ubiquitin pathways, variable biological features including variable expression of corticotropin-releasing hormone (CRH), glucocorticoid and dopaminergic receptors and variable proopiomelanocortin processing [17-20]. Rarely it occurs in patients with multiple endocrine neoplasia type 1 or 4, familial isolated pituitary adenoma, McCune-Albright syndrome or Carney complex [15, 16, 20].
CD may in some cases be difficult to identify in magnetic resonance imaging (MRI) studies, requiring invasive and technically demanding petrosal sinus catheterization, and can only be cured by surgical removal generally using the transphenoidal route, with a success rate of about 60-70% in the long term in the best series [15, 16, 21].
Despite a standardized clinical diagnostic and treatment approach [22-24], CD may be difficult to diagnose and to treat; and several uncommon variants are recognized like cyclic CD [25], intermediate lobe corticotroph adenomas [26], mixed corticotroph adenomas [27], silent corticotroph macroadenomas [28] and even the exceedingly rare corticotroph carcinoma [29]. Also apparent CD because of CRH hypersecretion, either a hypothalamic tumor or a peripheral neuroendocrine tumor [30], and the classic Nelson’ syndrome [31] after bilateral adrenalectomy must be considered. The relations of these variants to the biological features of the tumor are not completely understood.
We now report what seems to be a common variant of CD, with distinctive clinical, analytical and imagiological findings, recurring after pituitary surgery, which presents as a clinical challenge and may be informative of the pathogenesis of CD in general. We assume this variant is commonly recognized by other groups, although we could not find a formal description in the literature. Furthermore adaptation to chronic low-grade hypercortisolism is apparent.
| Case Report | ▴Top |
Investigations
The patient is a Caucasian female, aged 29, single, working as a practicing nurse in a public central hospital in Lisbon and living in the Setubal district, less than 50 km south of Lisbon.
Since 1 year before, the patient had complained of headaches. In fact these were old complaints and with the general characteristics of migraine, with a periodicity of about once every month outside the menstrual period, with weight gain of about 8 kg since she was aged 20, associated with carbohydrate craving during periods of job stress, with no overweight and without a central distribution pattern of body fat, and with body striae that were in fact very mild at the thighs, thin, white and more easily explained by the weight gain. The patient emphatically insisted she was always very tired and that her body shape was changing. There was no diabetes mellitus, nor symptoms like increased thirst and frequent urination. Blood pressure was normal without any medication or cardiovascular symptomatology; and menstrual cycles were regular under contraceptive pill that was interrupted 2 months before, without any complains about hirsutism or acne. There were no previous attempted pregnancies. No visual defects were apparent and no other neurologic complaints were expressed.
There was a past episode suggestive of acute pyelonephritis. Hypermetropia was corrected some years ago by ocular surgery. No other diseases or complaints were recorded.
The patient did not smoke, drank only socially (ethanol consumption < 40 g/week), and was not taking any regular medications.
Her father, aged 57 was healthy except for high blood pressure under medical treatment; and her mother, aged 56, had previous diagnosis of breast carcinoma and non-Hodgkin lymphoma, both cured. One older brother aged 35 was healthy. There was no family record for endocrine diseases, breast diseases or hematologic diseases in direct relatives.
Physical examination (Fig. 1) revealed an apparently healthy young female who was anxious, but otherwise with no behavioral alterations. Body temperature was normal (36 °C), conjunctives were not pale, and there was no peripheral edema. Height was 174 cm without shoes, weight was 69 kg (without shoes or coats), body mass index 22.8 kg/m2, umbilical perimeter 79 cm, waist perimeter 101 cm, thigh perimeter 62 cm (umbilical/waist ratio: 0.78); blood pressure 130/75 mm Hg in the right arm supine position, heart rate 80/min, and breath rate was 16 respirations/min. Head and neck examination were normal without alopecia, hirsutism, or acne lesions, no neurologic abnormalities regarding the cranial nerves on crude examination; and the thyroid was not enlarged and presented no palpable thyroid nodules. Chest examination revealed normally developed adult breasts with no nipple discharge and no cutaneous abnormalities, and breath and heart sound were supple, regular and with no abnormal findings. Abdominal examination revealed no tenderness, without any palpable masses and bowel sounds were normally present; there were no abdominal striae. Members examination did not revealed any changes with normal muscular tone, no evidence for venous abnormalities; and arterial pulses were present, regular and symmetrical; only minimal thin white striae could be found at the upper thighs. Crude neurologic examination was normal regarding muscle power, reflexes, motility, equilibrium or motor coordination.
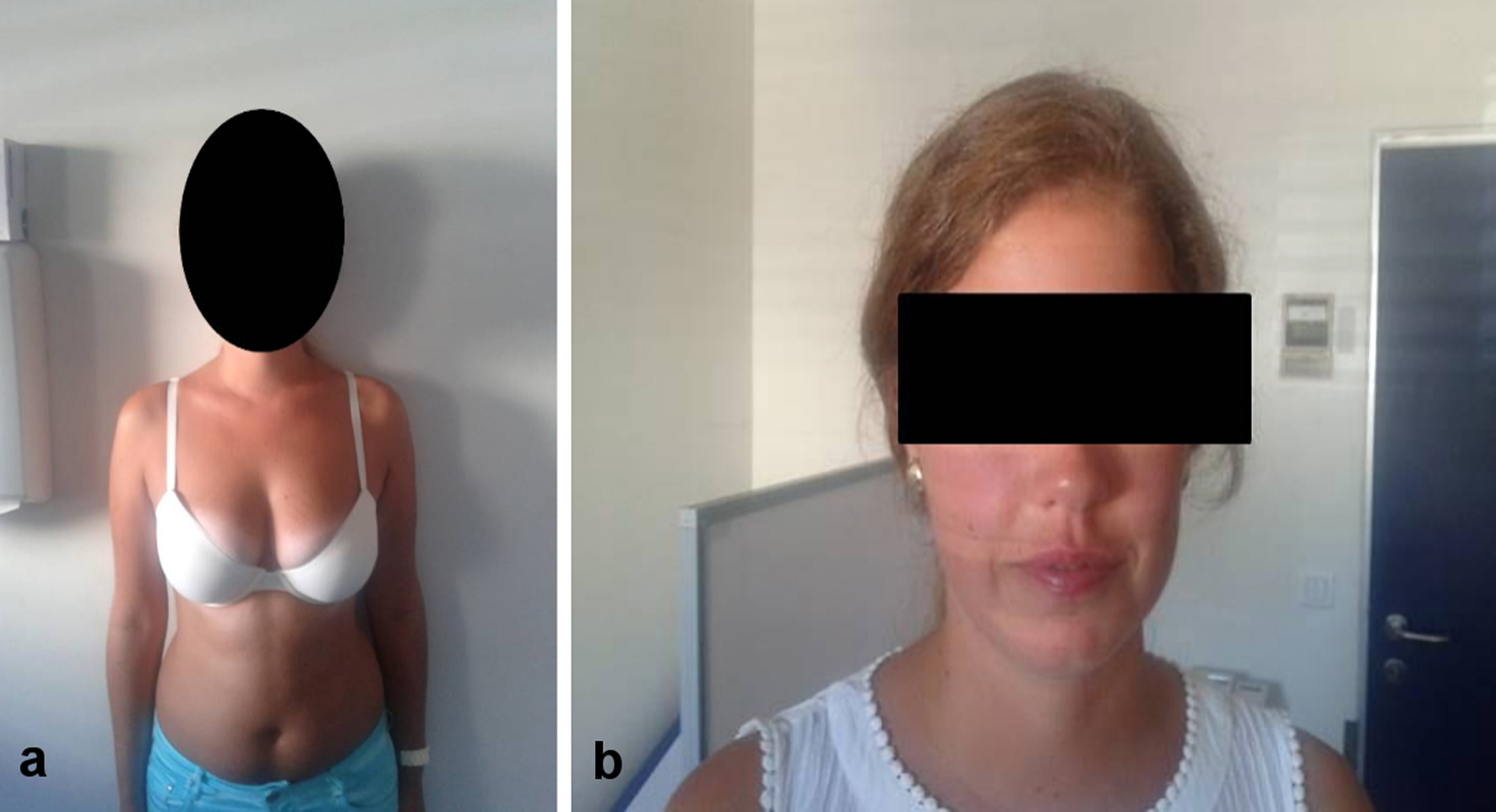 Click for large image | Figure 1. Physical habitus of patient. |
Diagnosis
Since she was a health professional working in a public central hospital in Lisbon, with easy access to medical care, a head MRI study had already been obtained that revealed a large intra-sellar mass with 19 × 13 mm, extending up until the optic nerves with no lateral invasion of the cavernous sinuses. That mass representing the whole pituitary gland was hypointense in both T1- and T2-weighted sections with homogenous uptake of the contrast. There was a widening of the pituitary stalk and the neurohypophysis could not be defined clearly (Fig. 2).
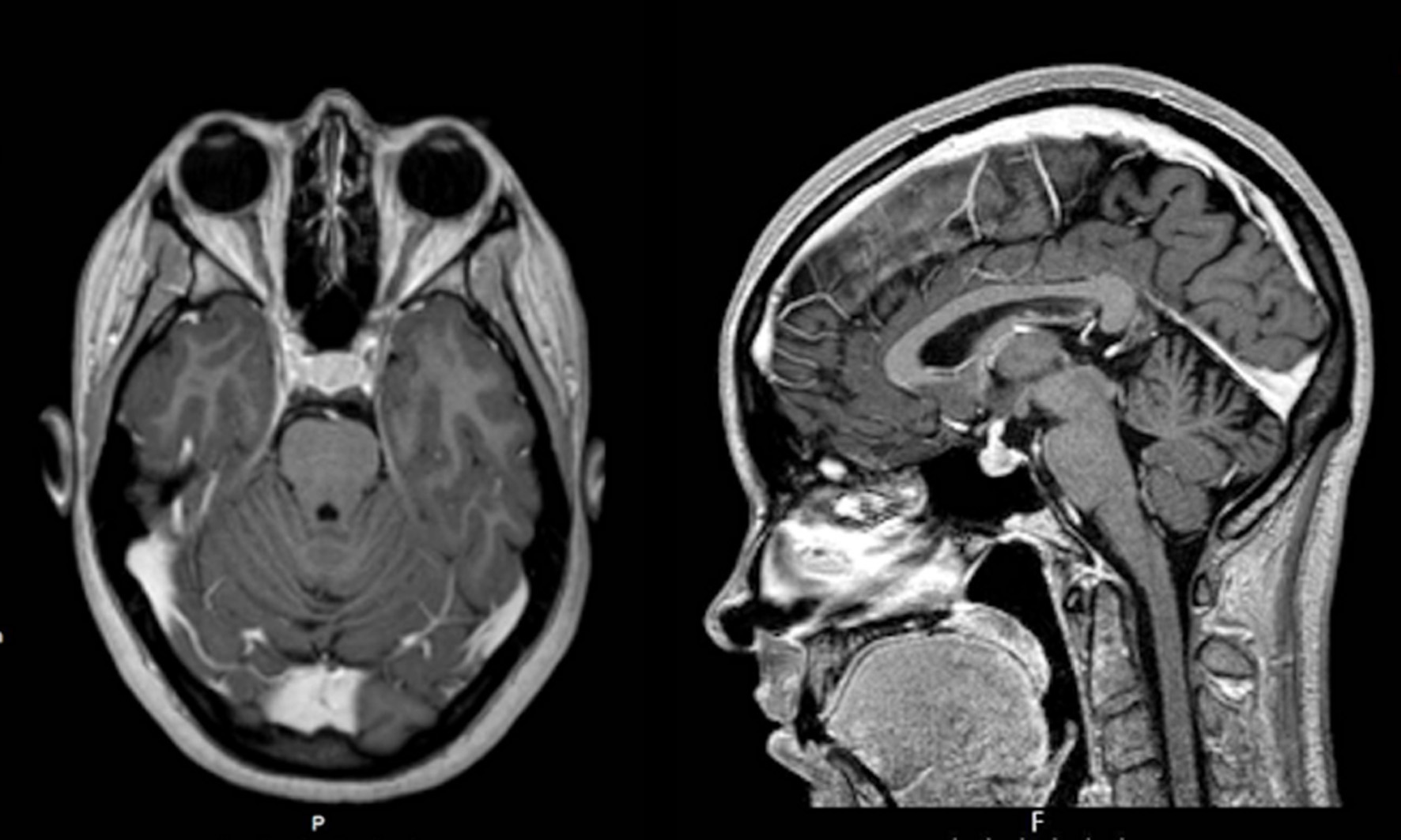 Click for large image | Figure 2. First MRI study showing diffuse enlargement of the whole pituitary gland. MRI: magnetic resonance imaging. |
Also at another institution a complete analytical blood panel had been obtained with no hematologic or biochemical routine abnormalities. However at 9:00 am, ACTH was 40 pg/mL (reference values (RV): 7 - 63), cortisol 25 µg/dL (RV: 10 - 20) and 24-h urinary cortisol 596 µg (RV: < 285). Normal values of growth hormone (GH), insulin-like growth factor 1 (IGF1), free T4 (FT4), thyroid-stimulating hormone (TSH), prolactin, follicle-stimulating hormone (FSH) and luteinizing hormone (LH) were found. An overnight dexamethasone test (1.0 mg orally (PO) at 24 h) had been performed with a cortisol value at 8:00 am next morning of 29 µg/dL. As an outpatient at that institution, a prolonged low-dose dexamethasone test (0.5 mg, every 6 h for 48 h) was obtained with a cortisol value of 39 µg/dL at 8:00 am after the last dose. A prolonged high-dose dexamethasone test (2 mg every 6 h for 48 h) test revealed a cortisol value of 2.9 µg/dL, 8:00 am after the last dose. Although done at another institution in the same city of Lisbon, the same analytical methods were used in our institution and are later described.
Since there was a marked discrepancy between clinical and analytical data, and the later were obtained on a non-supervised outpatient basis, we decided to repeat that evaluation as an inpatient.
Baseline routine analytical evaluation obtained in morning after the overnight fast revealed no change with normal hematological values without leukocytosis or eosinopenia, normal renal function with no electrolyte or mineral abnormalities, normal serum glucose, lipids and proteins, and normal hepatic enzymes. Chest X-ray and electrocardiogram (EKG) were normal. Baseline endocrine evaluation was normal as follows: T3: 80 ng/dL (RV: 80 - 200), T4: 6.4 ng/dL (RV: 5 - 14), TSH: 2.6 µU/mL (RV: 0.3 - 4.2), FT4: 1.22 ng/dL (RV: 0.9 - 1.7), thyroid peroxidase antibody (TPOAb): < 10 U/mL (RV: < 10), thyroglobulin antibody (TgAb): < 10 U/mL (RV: < 10), prolactin: 15 ng/mL (RV: 5 - 23), GH: 7.6 ng/mL (RV: < 10), IGF1: 431 ng/mL (RV: 117 - 329); IGFBP3: 5.8 ng/mL (RV: 3.5 - 7.6), FSH: 9 U/L (RV: 2.5 - 10), LH: 6 U/L (RV: 2 - 12.5), E2: 42 pg/mL (RV: 11 - 200), progesterone: 0.8 ng/mL (RV: 3.5 - 26), TT: 22 ng/dL (RV: 0 - 81), androstenedione: 2.7 ng/mL (RV: 0.3 - 3.3), sex hormone binding globulin (SHBG): 32 nmol/L (RV: 18 - 114); parathyroid hormone (PTH): 33 pg/mL (RV: 7 - 65); 25-hydroxyvitamin D: 22 ng/mL (RV: 20 - 80), insulin: 11 mU/mL (RV: 3 - 29); C-peptide: 1.7 ng/mL (RV: 1.1 - 5.0). Markers of bone remodeling were also normal or slightly increased: osteocalcin 14 ng/mL (RV: 3.1 - 13.7); N-terminal fragments of type 1 collagen propeptide (P1NP): 49 pg/mL (RV: < 30); C-terminal fragments of type 1 collagen (CTX): 0.45 ng/mL (RV: < 0.3). Nonspecific markers for neuroendocrine tumors were normal: chromogranin A: 1.9 (RV: < 3) and neuron-specific enolase: 21 (RV: < 18). Specifically and regarding the pituitary-adrenal axis the following parameters were obtained: ACTH: 48 pg/mL (RV: 7 - 63), cortisol: 20 µg/dL (RV: 10 - 20), dehydroepiandrosterone sulphate (DHEAS): 370 µg/dL (RV: 35 - 430); 17OHP: 2.2 ng/mL (RV: 0.2 - 4.7); S compound: 2.5 ng/mL (RV: < 8); aldosterone: 111 pg/mL (RV: 10 - 180); renin: 5.3 µU/mL (RV: 2 - 20), and 24-h urinary cortisol: 1,059 µg (RV: < 285).
All analytical measurements were obtained at the Clinical Pathology Department of the hospital using commercially available automated and standardized methods. Specifically electrochemoluminescence immunoassay (ECLIA) methods (Roche, Cobas 8000, Basel) were used for hormone measurements. Intra- and interassay variation coefficients were always below 10%. Reference values for the adult population are established by the Clinical Pathology Department and periodically revised to sustain clinical decisions. The Clinical Pathology Department is certified by the international standard ISO 9001:2015 and regularly participates in official quality control programs.
The circadian rhythm was evaluated followed by the rapid overnight dexamethasone (1.0 mg PO at 24 h) test (Table 1). Next, the prolonged low (dexamethasone 0.5 mg 6/6 h for 48 h) and high (dexamethasone 2 mg 6/6 h for 48 h) tests were obtained sequentially (Table 2). Finally a CRH test was obtained (hCRH 100 µg, intravenous (IV), bolus at time 0) (Table 3).
 Click to view | Table 1. Circadian Rhythm and Rapid Overnight Dexamethasone (1.0 mg PO at 24 h) Test |
Click to view | Table 2. Prolonged Low (Dexamethasone 0.5 mg 6/6 h for 48 h) and High (Dexamethasone 2 mg 6/6 h for 48 h) Tests |
Click to view | Table 3. CRH Test (hCRH 100 µg, IV, Bolus at Time 0) |
Abnormalities with possible pathogenic significance in the glucocorticoid receptor (GCR) coding gene (NR3C1) at 5q31 were not detected (exons and flanking introns) by QXT, Agilent Technologies and next generation sequencing MiSeq, Illumina with NextGENe, SoftGenetics (CGC Genetics, Porto, Portugal). CRH measurement at a private institution revealed low/suppressed CRH levels < 1 ng/mL (RV: 0 - 3.5) (enzymatic immunoassay (EIA), Cusabio®, Dr. Joaquim Chaves, Laboratorio de Analises Clinicas, Lisbon, Portugal).
Treatment
Six months later a pituitary adenoma was removed by sublabial incision and the transphenoidal route. Surgery was uneventful. Pathological examination (Fig. 3) revealed adenohypophysis and neurohypophysis fragments as well as fragments with the pattern of solid adenoma, some with eosinophilic and some with clear cytoplasm; no mitoses were found and anisokaryosis was mild to moderate; and Ki67 was less than 3%. Some cells were stained for ACTH by immunocytochemistry. On the fifth postsurgical day (8:00 am) the following values were obtained: ACTH 37 pg/mL and cortisol 9 µg/dL. Two weeks after surgery the values were as follows: ACTH 48 pg/mL and cortisol 25 µg/dL.
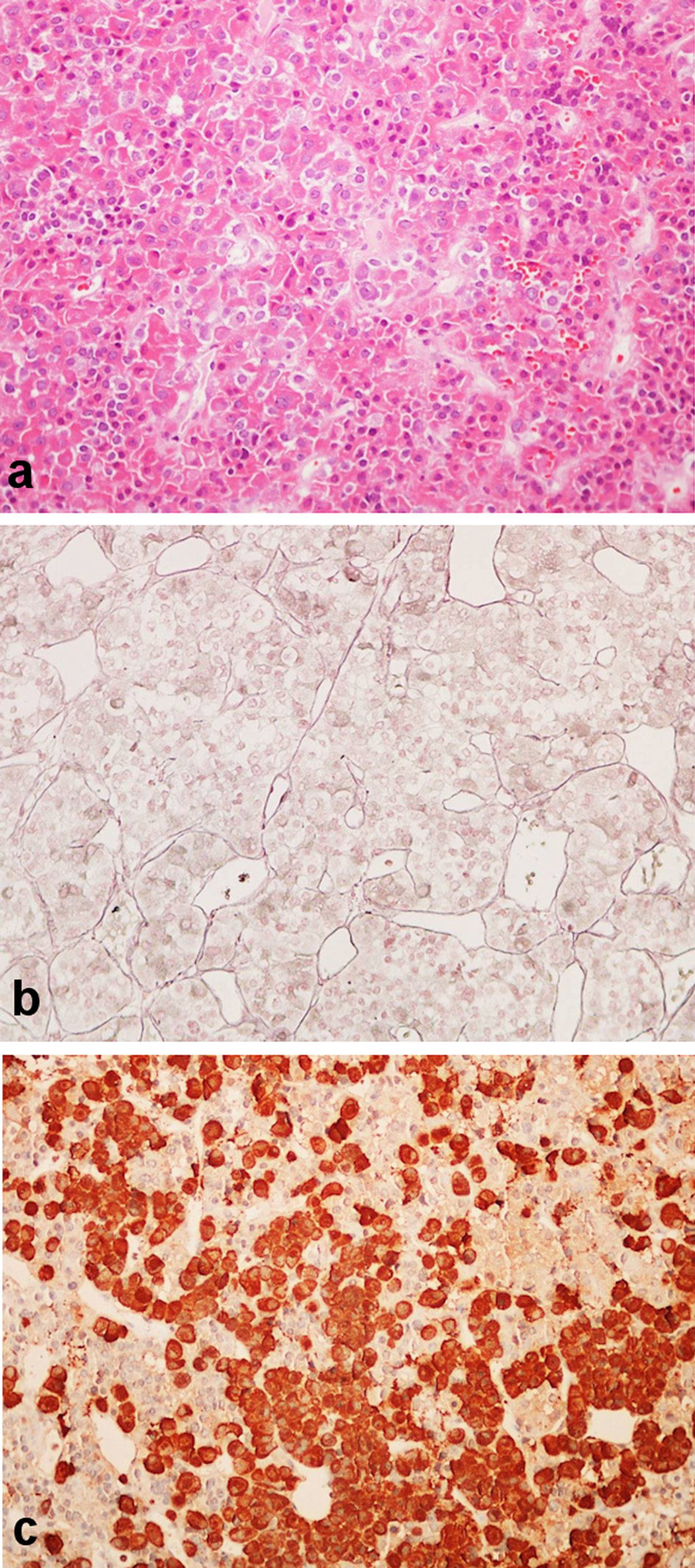 Click for large image | Figure 3. Pathologic sample removed at surgery. (a) Pituitary adenoma solid pattern, with cells with clear or eosinophilic cytoplasm (hematoxylin and eosin stain, × 200). (b) Scarcity of the reticulin net (Gomori stain × 200). (c) ACTH immunostain (× 200). ACTH: adrenocorticotropic hormone. |
Follow-up and outcomes
Six months later, under no medication, the patient maintained the same complaints as before, and menstrual cycles had not resumed after suspending the contraceptive pill. These complaints even if with poor objective translation had a profound impact on the quality of life of the patient. Analytical evaluation revealed ACTH: 44 pg/mL, cortisol: 17 µg/dL, 24-h urinary cortisol: 364 µg. Cortisol post dexamethasone (1.0 mg) the previous night: 15 µg/dL.
A new MRI did not reveal any pituitary abnormalities except for the persistent pituitary stalk widening (Fig. 4).
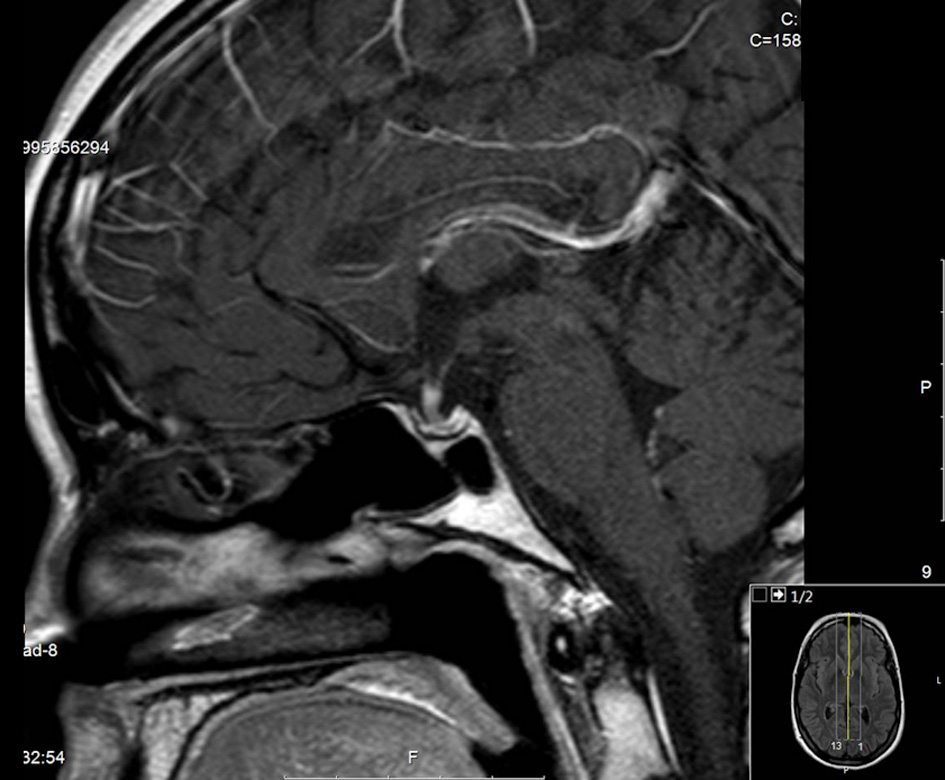 Click for large image | Figure 4. Last MRI study with marked widening of the pituitary stalk. MRI: magnetic resonance imaging. |
A head scan with the radiolabeled somatostatin analogue (111In-pentetreotide) was obtained with negative results; but despite that the somatostatin analogue (octreotide acetate, Sandostatin LAR®) 20 mg and later 30 mg, once a month, intramuscular (IM) was began and maintained for 8 months. Evaluation then found ACTH: 36 pg/mL; cortisol: 19 µg/dL; and 24-h urinary cortisol: 384 µg. Gallbladder stones developed although asymptomatic and less than 15 mm. Somatostatin was interrupted and cabergoline 0.5 mg, twice a week was began and maintained for 6 months: ACTH: 43 pg/mL; cortisol: 23 µg/dL; 24-h urinary cortisol: 158 µg; after 1 year ACTH: 44 pg/mL, cortisol: 17 µg/dL, 24-h urinary cortisol: 538 µg, and cortisol post dexamethasone (1.0 mg, PO) previous night: 27 µg/dL. Imipramine 25 mg, once daily at night was also attempted; and after 3 months it was revealed that ACTH: 72 pg/mL; cortisol: 16 µg/dL; 24-h urinary cortisol: 599 µg, and cortisol post dexamethasone (1.0 mg, PO): 14 µg/dL.
| Discussion | ▴Top |
This is indeed a very atypical case of CD. Despite patient insistence, there was no objective clinical evidence for hypercortisolism except for amenorrhea that could only later be defined and maybe emotional changes that are common in patients with CD [12, 16, 32, 33]. There was no central distribution of body fat or protein wasting namely proximal myopathy or purple wide skin striae that strongly characterizes hypercortisolism; there was no diabetes mellitus or glucose intolerance, no high blood pressure or dyslipidemia and no osteopenia or osteoporosis, no record for previous fractures or thrombotic events; there was no hirsutism or acne; there were no visual field defects, and headaches could be more easily explained by migraine [12, 16].
Despite lack of objective clinical evidence for hypercortisolism and again emphasizing the truism that “the patient is always right”, analytical evidence repeatedly and unequivocally revealed hypercortisolism (see 24-h urinary cortisol). Interestingly enough the circadian rhythm was almost maintained since values at 19 - 20 h are about half of those at 8 - 9 h, a very rare finding in CD; midnight cortisol values were however unequivocally increased [12, 16, 22, 23].
Furthermore and following a standardized diagnostic approach hypercortisolism was non suppressible by dexamethasone overnight; hypercortisolism was ACTH-dependent, and hypercortisolism was also not suppressed by the prolonged low-dose dexamethasone test, essentially excluding pseudo Cushing states like those of major depression and chronic stress [12, 16, 22, 23, 34, 35], but suppressed by the high-dose prolonged dexamethasone test, essentially excluding ectopic ACTH secretion [22, 23, 36].
New diagnostic approaches to the differential diagnosis of CS emphasize the central role of the CRH or desmopressin test [37]. In the CRH test, a 30-40% increase in ACTH and a 20% increase in cortisol are generally used to define CD; while in ectopic CS baseline ACTH and cortisol values are generally much higher and the response is much less; and in chronic CRH oversecretion a decreased ACTH response and increased cortisol response are described. By these criteria, in this case the CRH test suggests CD albeit imperfectly, and is strongly against the other possibilities [12, 16, 22, 23, 37-40].
Common pitfalls in the diagnosis of CD do not seem to explain discrepancies; free cortisol is increased as evaluated by the 24-h urinary cortisol excluding cortisol-binding globulin (CBG) increase induced by pill; no drugs were used that could increase the metabolism of dexamethasone or present anti-glucocorticoid effects [12, 16, 22, 23, 37].
A certain degree of peripheral cortisol resistance is certainly suggested by the lack of clinical features of hypercortisolism despite analytical hypercortisolism, but familial syndromes of cortisol resistance do not fit the pattern since there is no high blood pressure and furthermore no abnormalities were found in the GCR gene and plasma CRH levels were low. It seems more probable an adaptive response to maintained low grade hypercortisolism [41-46].
Imagiological findings are also discordant. In classic cases a small (5 mm) round hypodense (T1-weighted sections) lesion is found; it may be hyperdense in T2-weighted sections and may or not uptake the contrast [15, 16, 22, 47, 48]. Instead in this case a large (19 × 12 mm) lesion that seems to include all the pituitary with pituitary stalk widening, no clearly visible neurohypophysis bright spot and hypointense in both T1 and T2 is found with homogenous contrast uptake. This might suggest either corticotroph hyperplasia (although evidence for CRH oversecretion is lacking as noted), silent corticotroph macroadenomas (but there is clear analytical evidence for hypercortisolism) or mixed type corticotroph adenomas (but no evidence for other hormone secretion is found) [26-28, 30]. Most interestingly is the pituitary stalk widening; this finding has been reported in relation to granulomatous or immune-mediated pituitary diseases in regard with hypopituitary states [15, 16]. Given the global involvement of the pituitary it may instead in this case correspond to the enlargement of the pars tuberalis that surrounds the pituitary stalk.
Selective removal of the pituitary adenoma whenever possible is the only curative option in CD even if the benefits of that treatment have not been proven in mild or subclinical disease [16, 21, 24]. Therefore the justification for surgery in this case may be questioned on clinical grounds but it is clear given the size and suprasellar extension. Surgical mortality is between 1% and 2%, success rates are between 70% and 90% in the short term, although recurrences rates of 10-15% at 10 years are reported, and in the long term hypopituitarism may occur in up to 50% of the patients, mainly in cases of macroadenoma or repeated surgeries [16, 21, 24, 49].
Surgery was however ineffective, as could be appreciated early in the postoperative period [16, 24, 50-52]. At 6 months, there is clear evidence for disease persistence after surgery with marked non-suppressible hypercortisolism. Hypercortisolism was however apparently reduced to almost half, and this would favor corticotroph hyperplasia, or partial remotion of the adenoma [16]. Furthermore there is now no apparent abnormality of the pituitary except for the persistent pituitary stalk widening, so that presumably that widening at least contributes to the sustained autonomous hypercortisolism, even if a non-visible microadenoma cannot be excluded.
Pathologic findings are also atypical; an adenoma was found with no suggestion of hyperplasia, but eosinophilic or mixed type, instead of basophilic and ACTH-producing cells. Again a mixed corticotroph adenoma, a silent corticotroph macroadenoma or an intermediate lobe corticotroph adenoma remain possible but no evidence was found for other hormone secretion, clinically or analytical, or for a different proopiomelanocortin (POMC) processing, since analytical marked hypercortisolism is found [15, 16, 24-28].
There was no clear therapeutic option in this young, childless patient with minimal clinical evidence for hypercortisolism [16, 24, 53, 54]. Because of minimal clinical evidence of hypercortisolism and the desire for motherhood, pituitary surgery, adrenal surgery or radiotherapy either conventional or gamma-knife were not considered appropriate; because of minimal clinical evidence of hypercortisolism medical therapies directed to the adrenals with mitothane, methyrapone or ketoconazole were not considered appropriate also given the limited efficacy of those therapies with common side effects [16, 24, 53-55].
Medical therapy directed at the pituitary was therefore attempted. As is common in CD but unusual regarding other pituitary tumors, the octreoscan was negative; but despite this somatostatin analog therapy was attempted and unsuccessful, although the new analogs may be more selective for the somatostatin receptors type 5 and may be more effective [16, 24, 56, 57]. Dopaminergic therapy was attempted with cabergoline, and this seemed to be effective for some time at least, but an escape phenomenon was apparent later. Interestingly enough dopaminergic therapy has been reported as effective mainly in intermediate lobe corticotroph adenomas [16, 24, 58, 59]. New therapies directed to intracellular mediators of corticotroph growth and function are not yet clinically available [60].
At this point we do not have a definitive diagnosis and even more important we do not have a clear therapeutic option. We encouraged the patient to become pregnant, assuming that this would occur spontaneously, and that CD would not complicate pregnancy given the minimal clinical features, so that pituitary surgery could be again considered, that if directed to the stalk widening may later compromise fertility. Besides a variant of CD, we cannot exclude with certainty pituitary hyperplasia secondary to CRH oversecretion, but hyperplasia was not reported and eosinophil and mixed cells were found and CRH levels were normal/low, nor even less probably cortisol resistance even if high blood pressure is lacking and weight gain is present, no abnormalities in the GCR gene were found and again CRH levels were suppressed.
Learning points
In short we report one case of what seems a common variant of CD with several worth noting characteristics: 1) female gender and young age; 2) minimal clinical features of hypercortisolism; 3) evident analytical hypercortisolism albeit with maintained cortisol circadian rhythm; 4) no evidence for increased central CRH drive; 4) macroadenoma with marked pituitary stalk widening; 5) mixed eosinophilic and clear cell adenoma; 6) persistence of the disease after apparent a successful surgery; 7) partial and only temporary response to dopaminergic agents, and no response to the somatostatin analog with absent somatostatin receptors in the octreoscan; 8) furthermore, a certain degree of acquired adaptive cortisol resistance is evident, which explains lack or only mild clinical features and therefore the rarity of this clinical presentation.
We assume many other patients like this one have been found and recognized by other centers and we strongly encourage a specific variant of CD to be thus defined.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Written informed consent was obtained from the patient ABS regarding publication of this report including figures.
Author Contributions
JMM assisted the patient and wrote the manuscript; CMM thoroughly discussed the case and co-wrote the manuscript; JM thoroughly discussed the case, performed surgery and thoroughly discussed the manuscript; DLP thoroughly discussed the case, performed pathologic examination and thoroughly discussed the manuscript.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
| References | ▴Top |
- Broersen LH, Pereira AM, Jorgensen JO, Dekkers OM. Adrenal insufficiency in corticosteroids use: systematic review and meta-analysis. J Clin Endocrinol Metab. 2015;100(6):2171-2180.
doi pubmed - Ejaz S, Vassilopoulou-Sellin R, Busaidy NL, Hu MI, Waguespack SG, Jimenez C, Ying AK, et al. Cushing syndrome secondary to ectopic adrenocorticotropic hormone secretion: the University of Texas MD Anderson Cancer Center Experience. Cancer. 2011;117(19):4381-4389.
doi pubmed - Zhang C, Jin J, Xie J, Ye L, Su T, Jiang L, Zhou W, et al. The clinical features and molecular mechanisms of ACTH-secreting pancreatic neuroendocrine tumors. J Clin Endocrinol Metab. 2020;105(11):3449-3458.
doi pubmed - Rossi R, Tauchmanova L, Luciano A, Di Martino M, Battista C, Del Viscovo L, Nuzzo V, et al. Subclinical Cushing's syndrome in patients with adrenal incidentaloma: clinical and biochemical features. J Clin Endocrinol Metab. 2000;85(4):1440-1448.
doi pubmed - Terzolo M, Reimondo G, Chiodini I, Castello R, Giordano R, Ciccarelli E, Limone P, et al. Screening of Cushing's syndrome in outpatients with type 2 diabetes: results of a prospective multicentric study in Italy. J Clin Endocrinol Metab. 2012;97(10):3467-3475.
doi pubmed - Omura M, Saito J, Yamaguchi K, Kakuta Y, Nishikawa T. Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res. 2004;27(3):193-202.
doi pubmed - Chiodini I, Mascia ML, Muscarella S, Battista C, Minisola S, Arosio M, Santini SA, et al. Subclinical hypercortisolism among outpatients referred for osteoporosis. Ann Intern Med. 2007;147(8):541-548.
doi pubmed - Glintborg D, Henriksen JE, Andersen M, Hagen C, Hangaard J, Rasmussen PE, Schousboe K, et al. Prevalence of endocrine diseases and abnormal glucose tolerance tests in 340 Caucasian premenopausal women with hirsutism as the referral diagnosis. Fertil Steril. 2004;82(6):1570-1579.
doi pubmed - Leon-Justel A, Madrazo-Atutxa A, Alvarez-Rios AI, Infantes-Fontan R, Garcia-Arnes JA, Lillo-Munoz JA, Aulinas A, et al. A probabilistic model for Cushing's syndrome screening in at-risk populations: a prospective multicenter study. J Clin Endocrinol Metab. 2016;101(10):3747-3754.
doi pubmed - Lindholm J, Juul S, Jorgensen JO, Astrup J, Bjerre P, Feldt-Rasmussen U, Hagen C, et al. Incidence and late prognosis of cushing's syndrome: a population-based study. J Clin Endocrinol Metab. 2001;86(1):117-123.
doi pubmed - Stewart PM, Newell-Price JDC. Chapter 15. The adrenal cortex. In: Melmed S, Polonsky KS, Larsen PR, Kronenberg HM (eds). Williams Textbook of Endocrinology. 13th edition. Elsevier, Philadelphia. 2016. p. 490-555.
doi - Juszczak A, Morris DG, Grossman AB, Nieman LK. Chapter 13. Cushing's syndrome. In: Jameson JL, De Groot LJ, Kretser DM, Giudice LC, Grossman AB, Melmed S, Potts JT, et al. (eds). Endocrinology: Adult & Pediatric, 7th edition. Elsevier Saunders, Philadelphia; 2016. p. 227-255.
doi - Rubinstein G, Osswald A, Hoster E, Losa M, Elenkova A, Zacharieva S, Machado MC, et al. Time do diagnosis in Cushing's syndrome: a meta-analysis based on 5367 patients. J Clin Endocrinol Metab. 2020;105(3):e12-e23.
doi pubmed - Papakokkinou E, Olsson DS, Chantzichristos D, Dahlqvist P, Segerstedt E, Olsson T, Petersson M, et al. Excess morbidity persists in patients with Cushing's disease during long-term remission: a Swedish nationwide study. J Clin Endocrinol. 2020;105(8):2616-2624.
doi pubmed - Melmed S, Kleinberg D. Chapter 9. Pituitary masses and tumors. In: Melmed S, Polonsky KS, Larsen PR, Kronenberg HM (eds). Williams Textbook of Endocrinology. 13th edition. Elsevier, Philadelphia; 2016. p. 232-299.
doi - Bertagna X, Guignat L, Raux-Demay M-C, Guilhaume B, Girard F. Chapter 16. Cushing's disease. In: Melmed S (ed). The Pituitary, third edition. Elsevier Academic Press, London; 2011. p. 533-617.
doi - Gicquel C, Le Bouc Y, Luton JP, Girard F, Bertagna X. Monoclonality of corticotroph macroadenomas in Cushing's disease. J Clin Endocrinol Metab. 1992;75(2):472-475.
doi pubmed - Theodoropoulou M, Reincke M, Fassnacht M, Komada M. Decoding the genetic basis of Cushing's disease: USP8 in the spotlight. Eur J Endocrinol. 2015;173(4):M73-83.
doi pubmed - Cassarino MF, Sesta A, Pagliardini L, Losa M, Lasio G, Cavagnini F, Pecori Giraldi F. Proopiomelanocortin, glucocorticoid, and CRH receptor expression in human ACTH-secreting pituitary adenomas. Endocrine. 2017;55(3):853-860.
doi pubmed - Chasseloup F, Pankratz N, Lane J, Faucz FR, Keil MF, Chittiboina P, Kay DM, et al. Germline CDKN1B loss-of-function variants cause pediatric Cushing's disease with or without an MEN4 phenotype. J Clin Endocrinol Metab. 2020;105(6):1983-2005.
doi pubmed - Bertagna X. MANAGEMENT OF ENDOCRINE DISEASE: Can we cure Cushing's disease? A personal view. Eur J Endocrinol. 2018;178(5):R183-R200.
doi pubmed - Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, Montori VM. The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93(5):1526-1540.
doi pubmed - Galm BP, Qiao N, Klibanski A, Biller BMK, Tritos NA. Accuracy of laboratory tests for the diagnosis of Cushing syndrome. J Clin Endocrinol Metab. 2020;105(6):2081-2094.
doi pubmed - Nieman LK, Biller BM, Findling JW, Murad MH, Newell-Price J, Savage MO, Tabarin A, et al. Treatment of Cushing's syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2015;100(8):2807-2831.
doi pubmed - Meinardi JR, Wolffenbuttel BH, Dullaart RP. Cyclic Cushing's syndrome: a clinical challenge. Eur J Endocrinol. 2007;157(3):245-254.
doi pubmed - McNicol AM, Teasdale GM, Beastall GH. A study of corticotroph adenomas in Cushing's disease: no evidence of intermediate lobe origin. Clin Endocrinol (Oxf). 1986;24(6):715-722.
doi pubmed - Rajendran R, Naik S, Sandeman DD, Nasruddin AB. Pasireotide therapy in a rare and unusual case of plurihormonal pituitary macroadenoma. Endocrinol Diabetes Metab Case Rep. 2013;2013:130026.
doi pubmed - Petrossians P, Ronci N, Valdes Socin H, Kalife A, Stevenaert A, Bloch B, Tabarin A, et al. ACTH silent adenoma shrinking under cabergoline. Eur J Endocrinol. 2001;144(1):51-57.
doi pubmed - Heaney AP. Clinical review: Pituitary carcinoma: difficult diagnosis and treatment. J Clin Endocrinol Metab. 2011;96(12):3649-3660.
doi pubmed - Karageorgiadis AS, Papadakis GZ, Biro J, Keil MF, Lyssikatos C, Quezado MM, Merino M, et al. Ectopic adrenocorticotropic hormone and corticotropin-releasing hormone co-secreting tumors in children and adolescents causing cushing syndrome: a diagnostic dilemma and how to solve it. J Clin Endocrinol Metab. 2015;100(1):141-148.
doi pubmed - Barber TM, Adams E, Ansorge O, Byrne JV, Karavitaki N, Wass JA. Nelson's syndrome. Eur J Endocrinol. 2010;163(4):495-507.
doi pubmed - Haskett RF. Diagnostic categorization of psychiatric disturbance in Cushing's syndrome. Am J Psychiatry. 1985;142(8):911-916.
doi pubmed - Tirosh A, RaviPrakash H, Papadakis GZ, Tatsi C, Belyavskaya E, Charalampos L, Lodish MB, et al. Computerized analysis of brain MRI parameter dynamics in young patients with Cushing syndrome - A case-control study. J Clin Endocrinol Metab. 2020;105(5):e2069-e2077.
doi pubmed - Alwani RA, Schmit Jongbloed LW, de Jong FH, van der Lely AJ, de Herder WW, Feelders RA. Differentiating between Cushing's disease and pseudo-Cushing's syndrome: comparison of four tests. Eur J Endocrinol. 2014;170(4):477-486.
doi pubmed - Isidori AM, Kaltsas GA, Mohammed S, Morris DG, Jenkins P, Chew SL, Monson JP, et al. Discriminatory value of the low-dose dexamethasone suppression test in establishing the diagnosis and differential diagnosis of Cushing's syndrome. J Clin Endocrinol Metab. 2003;88(11):5299-5306.
doi pubmed - Salgado LR, Fragoso MC, Knoepfelmacher M, Machado MC, Domenice S, Pereira MA, de Mendonca BB. Ectopic ACTH syndrome: our experience with 25 cases. Eur J Endocrinol. 2006;155(5):725-733.
doi pubmed - Frete C, Corcuff J-B, Kuhn E, Salenave S, Gaye D, Young J, Chanson P, et al. Non-invasive diagnostic strategy in ACTH-dependent Cushing's syndrome. J Clin Endocrinol Metab. 2020;105(10):3273-3284.
doi pubmed - Newell-Price J, Morris DG, Drake WM, Korbonits M, Monson JP, Besser GM, Grossman AB. Optimal response criteria for the human CRH test in the differential diagnosis of ACTH-dependent Cushing's syndrome. J Clin Endocrinol Metab. 2002;87(4):1640-1645.
doi pubmed - Peters CJ, Storr HL, Grossman AB, Savage MO. The role of corticotrophin-releasing hormone in the diagnosis of Cushing's syndrome. Eur J Endocrinol. 2006;155:S93-S98.
doi - Ceccato F, Tizianel I, Vedolin CK, Boscaro M, Barbot M, Scaroni C. Human corticotropin-releasing hormone tests: 10 years of real-life experience in pituitary and adrenal disease. J Clin Endocrinol Metab. 2020;105(11):e3938-e3949.
doi pubmed - Chrousos GP, Vingerhoeds A, Brandon D, Eil C, Pugeat M, DeVroede M, Loriaux DL, et al. Primary cortisol resistance in man. A glucocorticoid receptor-mediated disease. J Clin Invest. 1982;69(6):1261-1269.
doi pubmed - Raef H, Baitei EY, Zou M, Shi Y. Genotype-phenotype correlation in a family with primary cortisol resistance: possible modulating effect of the ER22/23EK polymorphism. Eur J Endocrinol. 2008;158(4):577-582.
doi pubmed - Martins JM, Kastin AJ, Banks WA. Unidirectional specific and modulated brain to blood transport of corticotropin-releasing hormone. Neuroendocrinology. 1996;63(4):338-348.
doi pubmed - Martins JM, Banks WA, Kastin AJ. Acute modulation of active carrier-mediated brain-to-blood transport of corticotropin-releasing hormone. Am J Physiol. 1997;272(2 Pt 1):E312-319.
doi pubmed - Martins JM, Banks WA, Kastin AJ. Transport of CRH from mouse brain directly affects peripheral production of β-endorphin by the spleen. Am J Physiol. 1997;273:1083-1089.
doi pubmed - Martin Martins J, Vale S, Ferreira F, Fagundes MJ, Carmo I, Saldanha C, Martins ESJ. Plasma corticotropin releasing hormone during the feeling of induced emotions. Neuro Endocrinol Lett. 2010;31(2):250-255.
- Bonneville JF. Magnetic resonance imaging of pituitary tumors. Front Horm Res. 2016;45:97-120.
doi pubmed - Dogansen SC, Yalin GY, Tanrikulu S, Tekin S, Nizam N, Bilgic B, Sencer S, et al. Clinicopathological significance of baseline T2-weighted signal intensity in functional pituitary adenomas. Pituitary. 2018;21(4):347-354.
doi pubmed - Yamada S, Inoshita N, Fukuhara N, Yamaguchi-Okada M, Nishioka H, Takeshita A, Suzuki H, et al. Therapeutic outcomes in patients undergoing surgery after diagnosis of Cushing's disease: A single-center study. Endocr J. 2015;62(12):1115-1125.
doi pubmed - Ioachimescu AG. Prognostic factors of long-term remission after surgical treatment of Cushing's disease. Endocrinol Metab Clin North Am. 2018;47(2):335-347.
doi pubmed - Witek P, Zielinski G, Szamotulska K, Maksymowicz M, Kaminski G. Clinicopathological predictive factors in the early remission of corticotroph pituitary macroadenomas in a tertiary referral centre. Eur J Endocrinol. 2016;174(4):539-549.
doi pubmed - Ironside N, Chatain G, Asuzu D, Benzo S, Lodish M, Sharma S, Nieman L, et al. Earlier post-operative hypocortisolemia may predict durable remission from Cushing's disease. Eur J Endocrinol. 2018;178(3):255-263.
doi pubmed - Espinosa-de-Los-Monteros AL, Sosa-Eroza E, Espinosa E, Mendoza V, Arreola R, Mercado M. Long-term outcome of the different treatment alternatives for recurrent and persistent Cushing disease. Endocr Pract. 2017;23(7):759-767.
doi pubmed - Geer EB, Shafiq I, Gordon MB, Bonert V, Ayala A, Swerdloff RS, Katznelson L, et al. Biochemical control during long-term follow-up of 230 adult patients with cushing disease: a multicenter retrospective study. Endocr Pract. 2017;23(8):962-970.
doi pubmed - Verhelst JA, Trainer PJ, Howlett TA, Perry L, Rees LH, Grossman AB, Wass JA, et al. Short and long-term responses to metyrapone in the medical management of 91 patients with Cushing's syndrome. Clin Endocrinol (Oxf). 1991;35(2):169-178.
doi pubmed - Pedroncelli AM. Medical treatment of Cushing's disease: somatostatin analogues and pasireotide. Neuroendocrinology. 2010;92(Suppl 1):120-124.
doi pubmed - Petersenn S, Salgado LR, Schopohl J, Portocarrero-Ortiz L, Arnaldi G, Lacroix A, Scaroni C, et al. Long-term treatment of Cushing's disease with pasireotide: 5-year results from an open-label extension study of a Phase III trial. Endocrine. 2017;57(1):156-165.
doi pubmed - Nakhleh A, Saiegh L, Reut M, Ahmad MS, Pearl IW, Shechner C. Cabergoline treatment for recurrent Cushing's disease during pregnancy. Hormones (Athens). 2016;15(3):453-458.
doi pubmed - Ferriere A, Cortet C, Chanson P, Delemer B, Caron P, Chabre O, Reznik Y, et al. Cabergoline for Cushing's disease: a large retrospective multicenter study. Eur J Endocrinol. 2017;176(3):305-314.
doi pubmed - Zhang D, Damoiseaux R, Babayan L, Rivera-Meza EK, Yang Y, Bergsneider M, Wang MB, et al. Targeting Corticotroph HDAC and PI3-Kinase in Cushing disease. J Clin Endocrinol Metab. 2021;106(1):e232-e246.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.