

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website https://www.jofem.org |
Case Report
Volume 11, Number 2, April 2021, pages 56-64
Clinical Variability of Hypophosphatasia in Colombian Patients: Case Reports
Ana Maria Zarante Bahamona, b, h, Juan Carlos Prieto Riverac, h, Jorge Rojasc, Vladimir Gonzalez Lopezd, Victor Vargasa, e, Melissa Rosero Arevalof, g
aInstituto Roosevelt, Bogota, Colombia
bLaboratorio Biotecnologia y Genetica, Bogota, Colombia
cPontificia Universidad Javeriana, Bogota, Colombia
dSubred Integrada de Servicios de Salud Suroccidente - Secretaria Distrital de Salud de Bogota, Colombia
eHospital Infantil Universitario de San Jose, Bogota, Colombia
fFundacion Clinica Infantil Club Noel, Cali, Colombia
gClinica Imbanaco, Cali, Colombia
hCorresponding Author: Ana Maria Zarante Bahamon, Instituto Roosevelt, Bogota, Colombia; Juan Carlos Prieto Rivera, Pontificia Universidad Javeriana, Bogota, Colombia
Manuscript submitted November 2, 2020, accepted November 9, 2020, published online April 25, 2021
Short title: Clinical Variability of HPP in Colombians
doi: https://doi.org/10.14740/jem711
| Abstract | ▴Top |
Hypophosphatasia (HPP) is a rare inherited disorder characterized by low serum alkaline phosphatase. It affects bone and tooth mineralization, although extra-skeletal manifestations are frequent. HPP is caused by loss-of-function mutations in the ALPL gene, encoding the protein tissue-nonspecific alkaline phosphatase. The phenotype is broadly variable, from a subtype with exclusive odontological compromise (odontohypophosphatasia) to five subtypes with systemic involvement, classified according to the age of onset at first symptoms. We present seven cases of HPP, in order to perform the clinical, biochemistry and radiological description of these Colombian patients, as well as to show the clinical variability of the disease in patients who present the same mutation or genetic defect.
Keywords: Short stature; Skeletal deformities; Alkaline phosphatase; Mutation; Hypomineralization
| Introduction | ▴Top |
Hypophosphatasia (HPP; OMIM #241500, 241510, 146300) is a rare and hereditary disorder of bone and mineral metabolism, first described by Rathbun in 1948, who reported low levels of alkaline phosphatase (ALP) activity in blood and in several tissues from an infant who died with rickets and epilepsy [1, 2]. It is caused by loss-of-function mutations in the tissue-nonspecific ALP (TNSALP) gene, the gene encoding the isoenzyme TNSALP, which is located in the chromosome 1p36.1-1p34. It is found predominantly in the bone, teeth, kidney and liver, but also in other cell types. This enzyme is located on the extracellular surface of the membrane and hydrolyses phosphates. In HPP, the reduced activity of TNSALP leads to the extracellular accumulation of inorganic pyrophosphate (PPi), pyridoxal 5′-phosphate (PLP) and phosphoethanolamine (PEA). The increased extracellular PPi acts as an inhibitor of skeletal mineralization [2] and it is the main metabolite associated with bone involvement [3-5].
HPP has been classified based on age at first manifestation of the disease, and the clinical presentation of HPP is influenced by mode of inheritance. There are six types of HPP described based on onset age: perinatal lethal, prenatal benign (in utero and at birth), infantile (age < 6 months), childhood (age ≥ 6 months to < 18 years), adult (age ≥ 18 years) and odontohypophosphatasia (this is the least severe form of HPP and affects only the teeth (spontaneous loss of fully rooted deciduous teeth or severe caries)) [6]. The large variation in clinical manifestations of the disease reflects the extent of the loss of enzyme function and may be indicated by a variety of symptoms and signs over a wide range of ages. In some cases, clinical manifestation or severity of the disease can be correlated with residual ALP activity and/or dominant negative effects [4, 5, 7].
The diagnosis of HPP is based on low level of serum ALP activity and genetic testing of the ALPL gene mutations. To date, approximately 410 mutations have been reported until May of 2020 (http://www.sesep.uvsq.fr/03_hypo_mutations.php) with the majority being missense variants which could explain the large heterogeneity in the clinical presentation and severity of the disease among affected individuals. However, the imperfect genotype-phenotype correlation has been observed, suggesting that other genetic or environmental factors modulate the phenotype and it makes genetic counselling and prognosis difficult [3, 7].
Since 2015, asfotase alfa (Strensiq®; Alexion Pharmaceuticals, Inc., New Haven, CT, USA), a human, recombinant TNSALP replacement therapy, replaces deficient TNSALP activity in patients with HPP and decreases the accumulation of substrates. It is available in different countries, given a new option of treatment for patients with HPP. Nowadays, Asfotase alfa was approved by Food and Drug Administration (FDA) and European Medicines Agency (EMA) for use in pediatric-onset HPP [4].
We present seven cases of unrelated Colombian patients with different types of HPP in order to perform the clinical, biochemical and radiological description of these Colombian patients, as well as to show the clinical variability of the disease in patients who present the same mutation and the different modes of inheritance.
| Case Reports | ▴Top |
The clinical phenotypes and biochemical test of all seven patients with HPP (six males and one female) are from seven unrelated families. Also, in order to confirm the diagnosis of HPP and try to correlate the phenotype with a specific genotype, mutational analyses of the ALPL gene were performed in all patients (Tables 1 and 2).
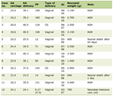 Click to view | Table 1. Laboratory Profile of Hypophosphatasia Patients |
Click to view | Table 2. Clinical, Molecular and Radiographic Findings |
Case 1
A male infant, product of second pregnancy, was born by C-section at 36 weeks with an antenatal history of short long bones and clinical suspicion of imperfect osteogenesis. There was no history of consanguinity. Radiographic studies showed alteration of the density and morphology of long bones from upper and lower limbs, decreased cortical bone, increased trabeculation and angulation of the diaphysis and focal bone defects of the metaphysis resembling radiolucent “tongues” (Figs. 1, 2).
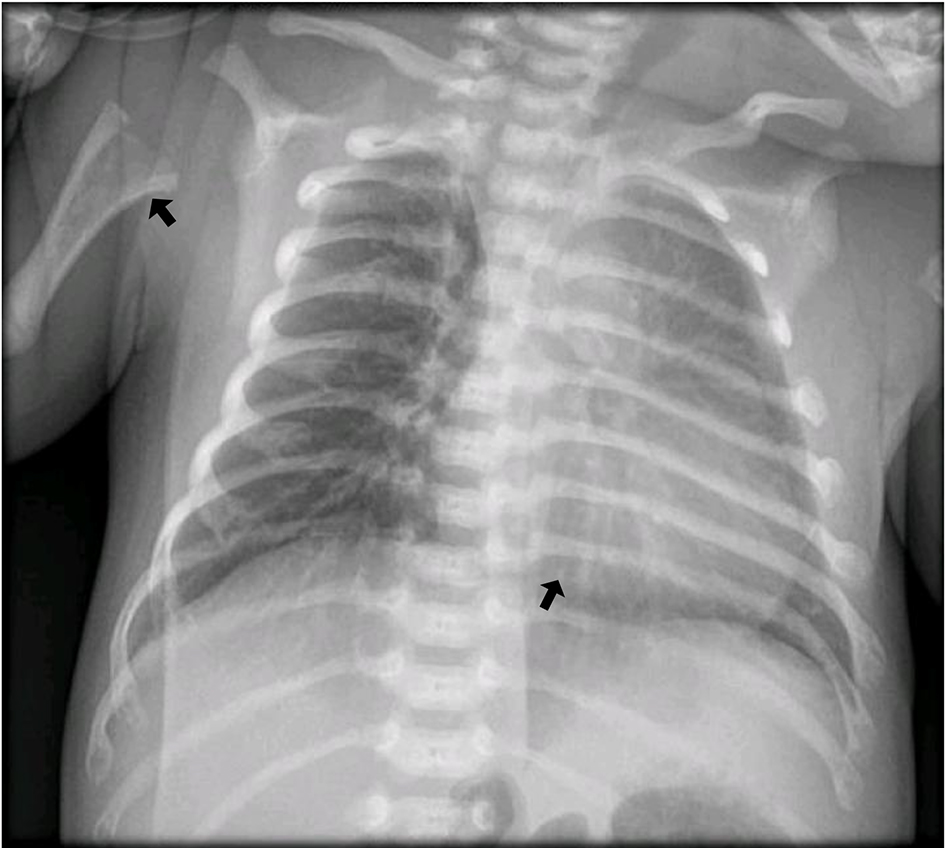 Click for large image | Figure 1. Horizontalization of the rib arches, decreased bone mineral density and slight thickening of the chondrocostal junctions (arrows). |
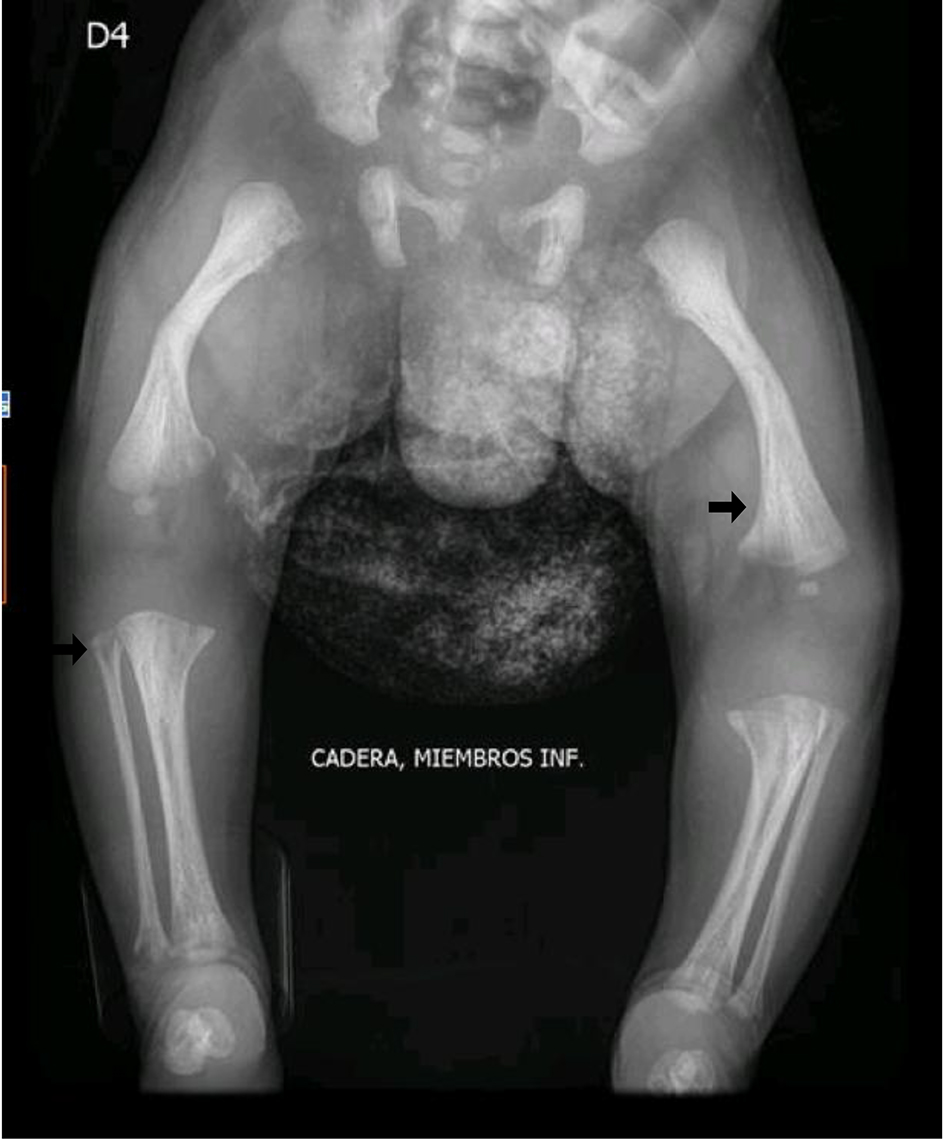 Click for large image | Figure 2. Before the treatment: alteration in the morphology of femurs with angulation of the diaphysis, mineralization defects in the bilateral metaphyses of the femurs, tibiae and fibula; alteration in the density and morphology of the long bones of the lower limbs (arrows). |
The physical examination at 6 months showed a height of 58.5 cm (-2.5 standard deviation (SD)), weight of 5.4 kg (-1.89 SD) and a head circumference of 40 cm (-2.5 SD). The following positive findings were observed: microcephaly, short neck, mild rhizomelic shortening and asymmetry of lower limbs.
Laboratory investigations revealed a serum calcium concentration of 9.5 mg/dL, a phosphate concentration of 8.6 mg/dL, an ALP level below 20 U/L (reference value: 134 - 518 U/L), parathyroid hormone (PTH) concentration of 19.2 pg/mL and a 25-hydroxyvitamin D concentration of 22.7 nmol/L. The neonatal form of HPP was diagnosed. Molecular study for ALPL reported a pathogenic variant c.892G>A (p.Glu298Lys) in a homozygous state.
Patient started treatment with asfotase alfa at 2 mg/kg of body weight administered subcutaneously three times per week, with adequate tolerance. He has not presented adverse effects and had adequate psychomotor development, improvement in height and weight for age, and bone mineralization (Fig. 3).
 Click for large image | Figure 3. After 3 months of treatment with asfotase. A decrease in the angulation of the diaphysis of the long bones is observed, the “tongue” defects disappear and there is an increase in bone mineralization and a decrease in metaphyseal widening (arrow). |
Case 2
A 26-year-old male patient with clinical and molecular diagnosis of childhood HPP with a marked decrease in ALP of 9.0 U/L (reference value: 40 - 129 U/L) arrived for clinical judgment. On complete sequencing of the ALPL, two pathogenic variants were identified: c.394G>A (p.Ala132Thr) and c.571G>A (p.Glu191Lys). The results were consistent with a diagnosis of autosomal-recessive HPP.
He had a disproportionate pathological short stature, osteomalacia and progressive bone deformities detected since age 3, history of pathological fractures in both arms and in spine with minimal traumas and premature loss of deciduous teeth at 5 years of age. Currently, he has varum deformities in the lower limbs, in which several corrective surgeries were performed and enamel hypoplasia of teeth was observed (Fig. 4).
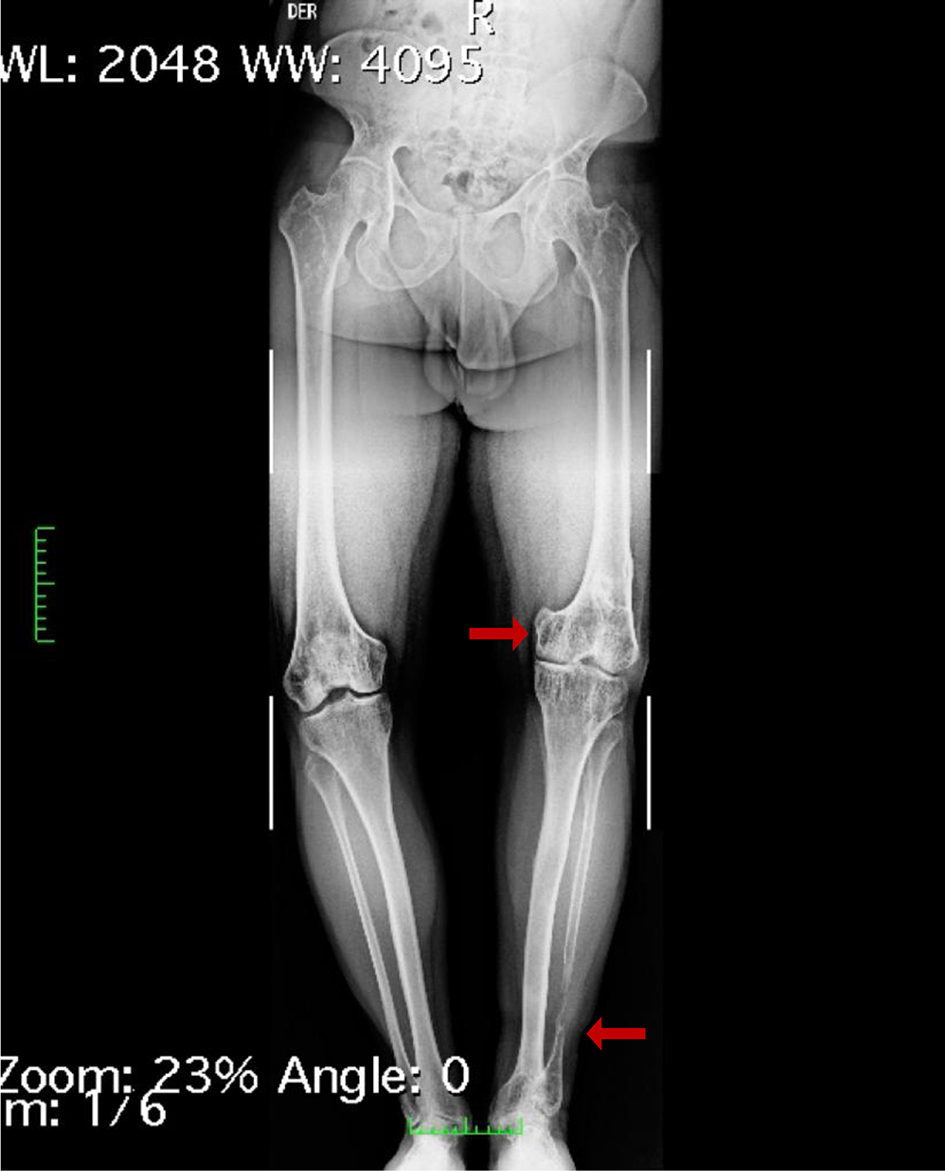 Click for large image | Figure 4. Genu varum, shortening of the left lower limb, loss of bone substance from the distal third of the left fibula, change of osteoarthritis in the knees and at the level of the neck of the left foot and marked osteopenia (arrows). |
He was assessed by nephrology who found nephrolithiasis and vitamin D deficiency. For this, endocrinology initiated supplementation with vitamin D 8,000 IU/week.
On physical exam, he had disproportionate short stature, asymmetric limbs, bowing and mesomelic shortening of the four limbs, asymmetry of lower limbs with genu varum, right radius and cubitus arching, brachydactyly at hands and feet, and diminished arc of movement in elbows.
Case 3
A 3-year 10-month-old male patient was referred to pediatric endocrinology by short stature and history of craniosynostosis (Fig. 5), without other relevant medical history. Physical exam showed a height of 92 cm (-2.16), weight of 13 kg (-2.39), body mass index (BMI) of 13.5 (-1.47), teeth with multiple damages and slight hyperflexibility in the upper extremities. No other relevant findings were noted.
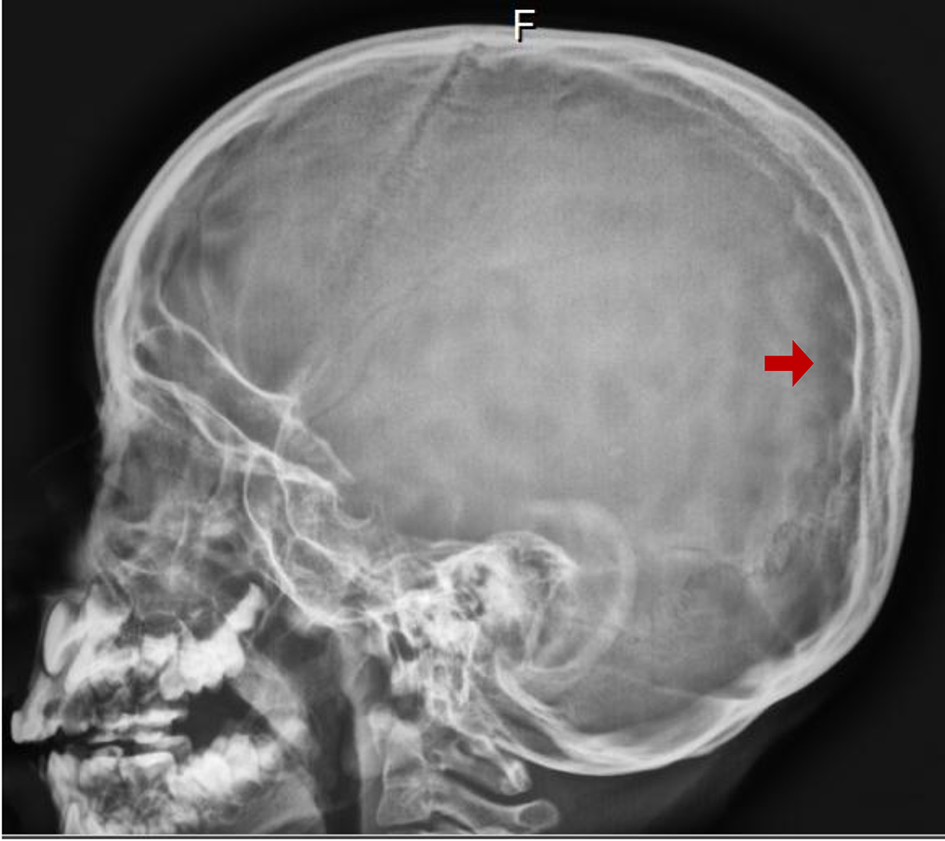 Click for large image | Figure 5. Lateral cranial X-ray: craneosynostosis is observed with a beaten silver appearance of the skull (arrow). |
Studies showed thyroid-stimulating hormone (TSH) of 2.68 µIU/mL, T4 of 1.8 ng/dL and insulin-like growth factor 1 (IGF-1) of 69 ng/mL. On carpogram, an “unusual” pattern of ossification was reported. For this reason, skeletal dysplasia, collagenopathies or HPP were proposed, and a complete phosphocalcic metabolic panel and referral to genetics were ordered.
Patient was seen by another pediatric endocrinologist and geneticist, both reporting laboratory findings as normal, diagnosing constitutional delay of growth and giving nutrition indications.
Patient returned at age 5 years 6 months with a height of 101.7 cm (-2.14), weight of 14 kg (-2.96), BMI of 13.5 (-2.02) and growth velocity of 4.3 cm/year (P < 3).
Laboratory profile showed 24-h calciuria of 1.5 mg/kg/day, phosphate tubular reabsorption of 94%, PTH of 24.07 pg/mL, 25OH-vitamin D of 53.24 ng/mL and ALP of 116.66 U/L (reference value: 156 - 369 U/L). Due to physical exam, impaired growth and laboratory findings, HPP was suspected. ALPL gene sequencing reported c.892G>A (p.Glu298Lys) in heterozygous state. The patient’s family was tested, showing father’s ALP of 122 U/L (reference value: 40 - 150 U/L) and mother’s ALP of 41 U/L (reference value: 40 - 150 U/L). The molecular test confirmed that mutation was inherited from his mother.
A comparative bone age study by Greulich and Pyle method corresponded to 2 years and 8 months with a patient’s chronological age of 4 years and 8 months. An “unusual” pattern of ossification was observed.
Case 4
A 12-year-old male patient with a history of delayed psychomotor development was referred for evaluation. At the age of 18 months, he went to orthopedics for gait delay, and premature loss of three teeth. Osteogenesis imperfecta was suspected. He was assessed by a geneticist in the United States who discarded it, and medical assessment was not continued. He walked after 18 months of age, and family members observed he presented impaired gait with easy fatigue, unable to do physical activity or walk for more than five blocks without pain, and progressive deformity in valgus in knees and ankles.
Physical exam showed low weight and height for age, gait with altered pattern, greater proximal effort, with difficulty for tiptoes, imbalanced gait, pain to palpation of the anterior superior iliac spine on the left side, brush and patellofemoral crepitation in both knees, increased external rotation of both tibiae and femurs, preserved hip rotations, severe knee pain with flexion above 90°, ankle with slight valgus, slight lumbar deviation to the right and major muscle retractions.
An X-ray and a magnetic resonance imaging (MRI) of both knees and a panoramic X-ray of lower limbs showed tongue lesions at the level of bilateral femur and tibia metaphysis, valgus ankle and knee deformity (Fig. 6). Bone hypomineralization metabolic disorder was suspected.
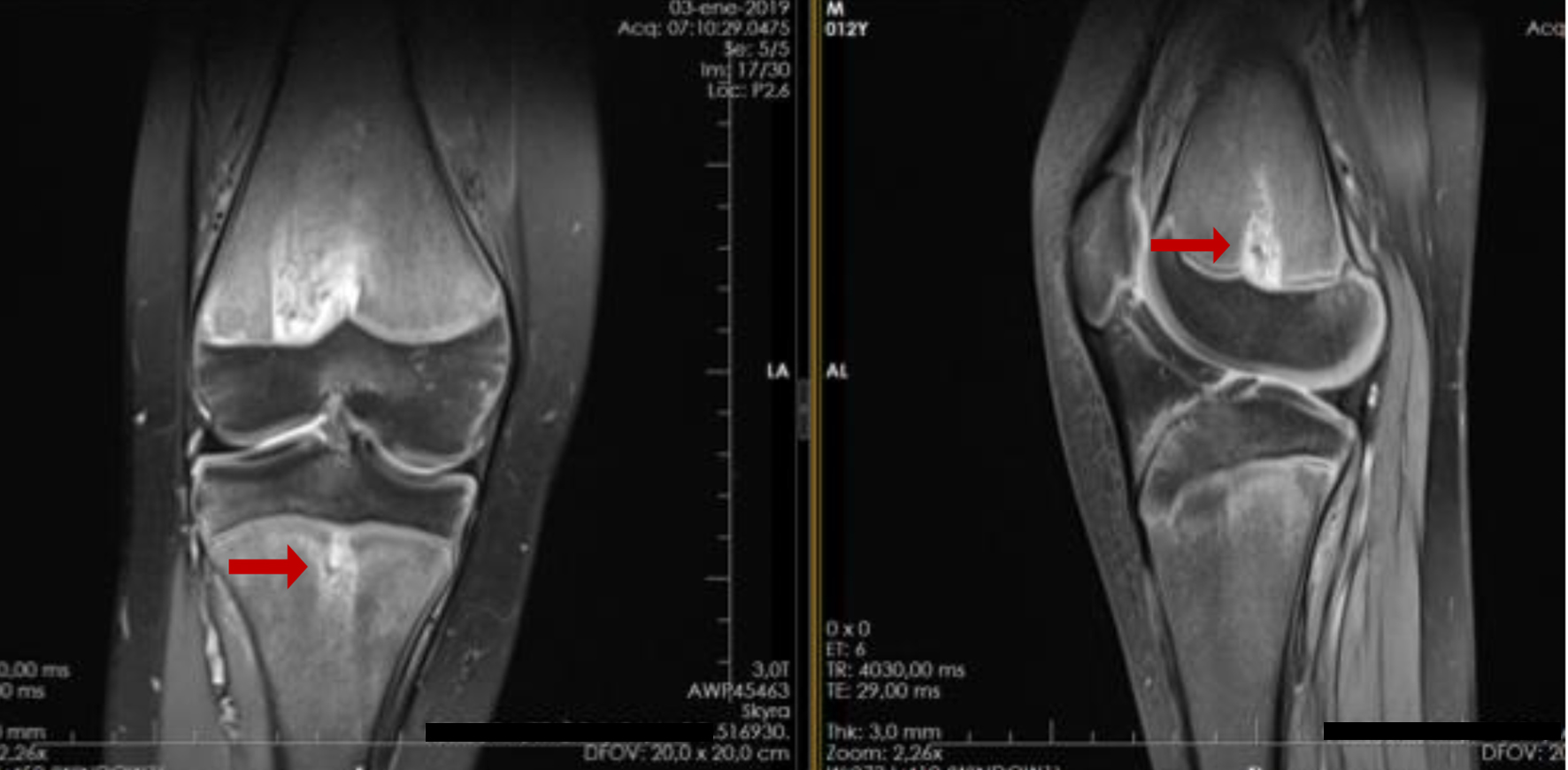 Click for large image | Figure 6. Magnetic resonance imaging of both knees: characteristic metaphyseal images on tongue (arrows). |
Laboratory tests showed ALP of 114 U/L (reference value: 141 - 460 U/L), creatine phosphokinase (CPK) of 97, phosphorus of 5.75 mg/dL, lactic dehydrogenase of 170 and calcium of 9.3.
Molecular analysis reported a likely pathogenic variant in heterozygous state c.334G>C (p.Gly112Arg) in ALPL. Parents were not studied, because they were not available.
Enzyme replacement therapy with asfotase alfa was started. At the end of the first month of therapy, the patient felt much better, with significantly lower pain in the limbs, and lower fatigue. Clinically, improvement in gait and knee range of motion was observed. By 3 months, he had significant improvement in gait, no longer tired so easily, improvement in muscle strength, achieved long-distance walks (several kilometers) and already rode a bicycle without getting tired easily; gait looked more stable, improvement of the knee valgus and full knee range of motion were noted.
Case 5
A 13-year-old boy had gait delay with premature loss of deciduous teeth (2 years), with family history of low levels of ALP (mother and sister) and parental consanguinity.
In the physical exam, there was evidence of a lame gait secondary to discrepancy of length in the lower limbs (shortening of the lower left limb), femoral retroversion, bilateral genu valgum and internal tibial torsion (Fig. 7). Radiological findings were typical, such as generalized osteopenia, femoral arching, and widening and areas of metaphyseal translucent. In addition, spondylodysplasia and pectus carinatum were noted.
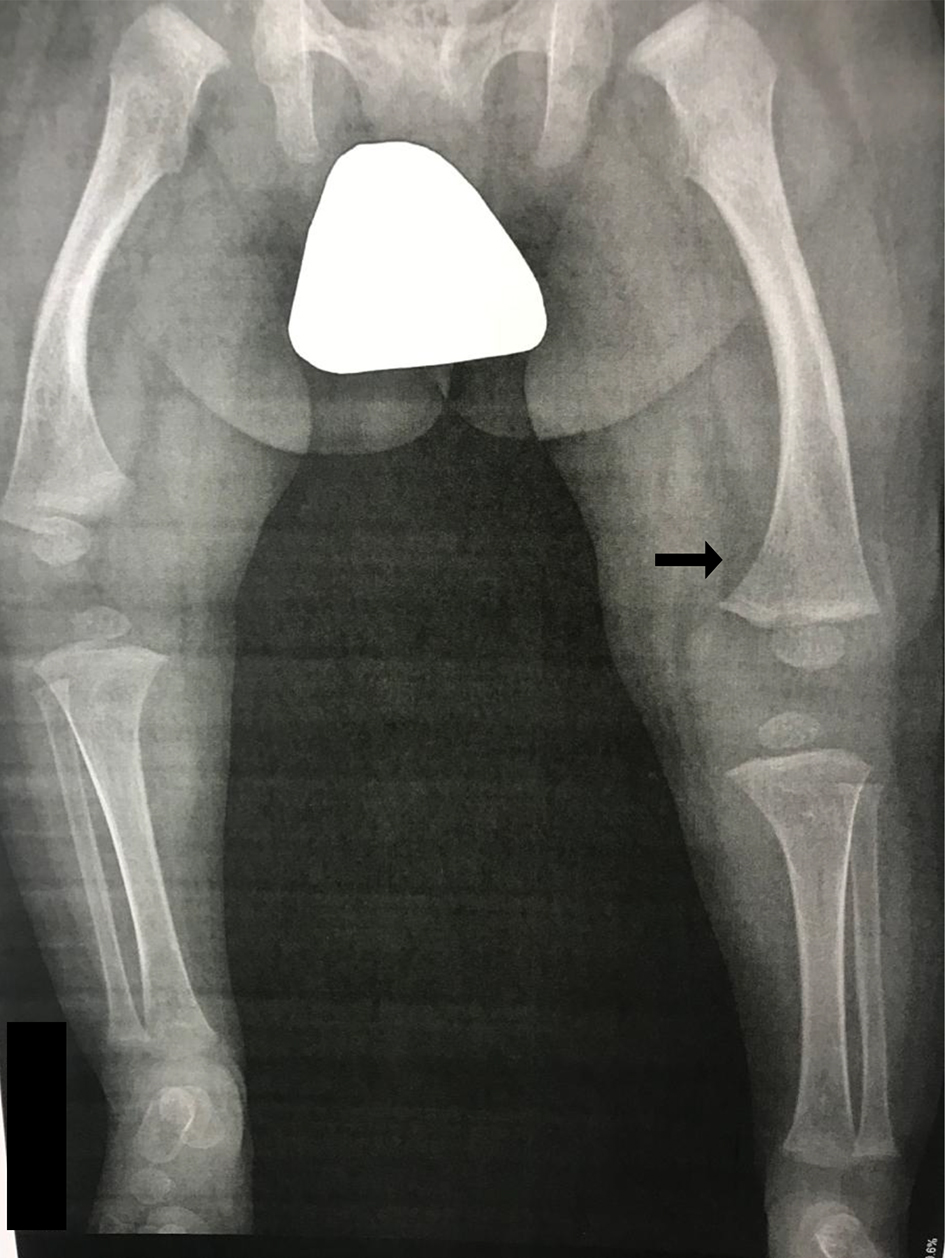
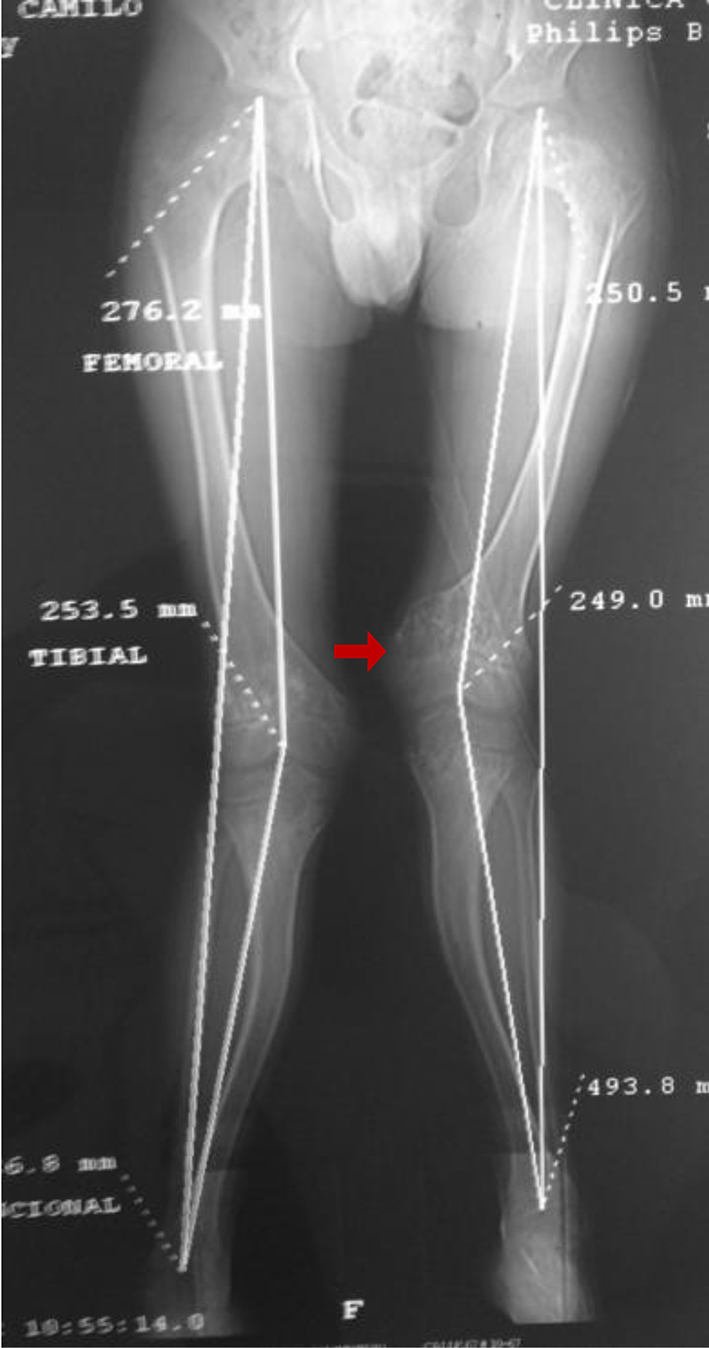 Click for large image | Figure 7. Bilateral genu valgum with multiapical deformities in the femur and tibia (arrow). |
ALP was low at 31 U/L (reference value: 141 - 460 U/L). The diagnosis of HPP was confirmed, and the sequencing of the ALPL gene reported homozygous for the mutation c.892G>A (p.Glu298Lys).
Surgical management was performed with left femoral and tibial osteotomies with endomedular fixation with telescoped nails and after right femoral and tibial osteotomies with endomedular fixation with telescoped nails (telescopes) and bilateral medial distal femoral hemiepiphysiodesis.
Currently, he presents an adequate clinical and functional evolution, and receives enzyme replacement therapy. He was asymptomatic, with gait without external aids, sustained growth and progressive improvement of angular alterations in his lower extremities.
Radiographic control studies of consolidated osteotomies, position of the appropriate osteosynthesis material, telescoping endomedular nails and the mechanical axis of both lower extremities have progressed towards normality.
Case 6
A 14-year-old male patient, second pregnancy product without complications, non-consanguineous parents, was evaluated.
At the age of 10 months, he had an obvious short stature that was associated at 15 months of age with chest deformity, frontal bossing and progressive genu valgum. In addition, he coursed with delayed standing and independent walking. At 2 years of life, non-traumatic tooth loss began. The patient started with seizures at 15 months of age until 3 years. No new seizures episodes were noted so far.
Finally, at 4 years of age, he was assessed by genetics that requested studies. Those evidence levels of ALP were low for age: 30 IU/L (reference value: 156 - 369 IU/L). At that time, a diagnosis of juvenile HPP was given; however, there was no therapy available and symptomatic management was given.
In 2013, patient was assessed again by genetics, showing progression of deformities. Long bone radiographs showed marked osteopenia of all bone structures, most at the proximal metaphysis of the femur, proximal and distal tibiae, distal metaphysis of the cubitus and radius, and proximal humeral metaphysis. Medial bowing of tibia and fibula, irregularity in the heads of the fibulae, osteopenia of bones of hands, universal widening and shortening of the costal arches (Fig. 8), varus deformity of the femoral necks and marked osteopenia of the pelvic bone structures and scoliosis were noted. ALP was 27 U/L (reference value: 141 - 460 U/L).
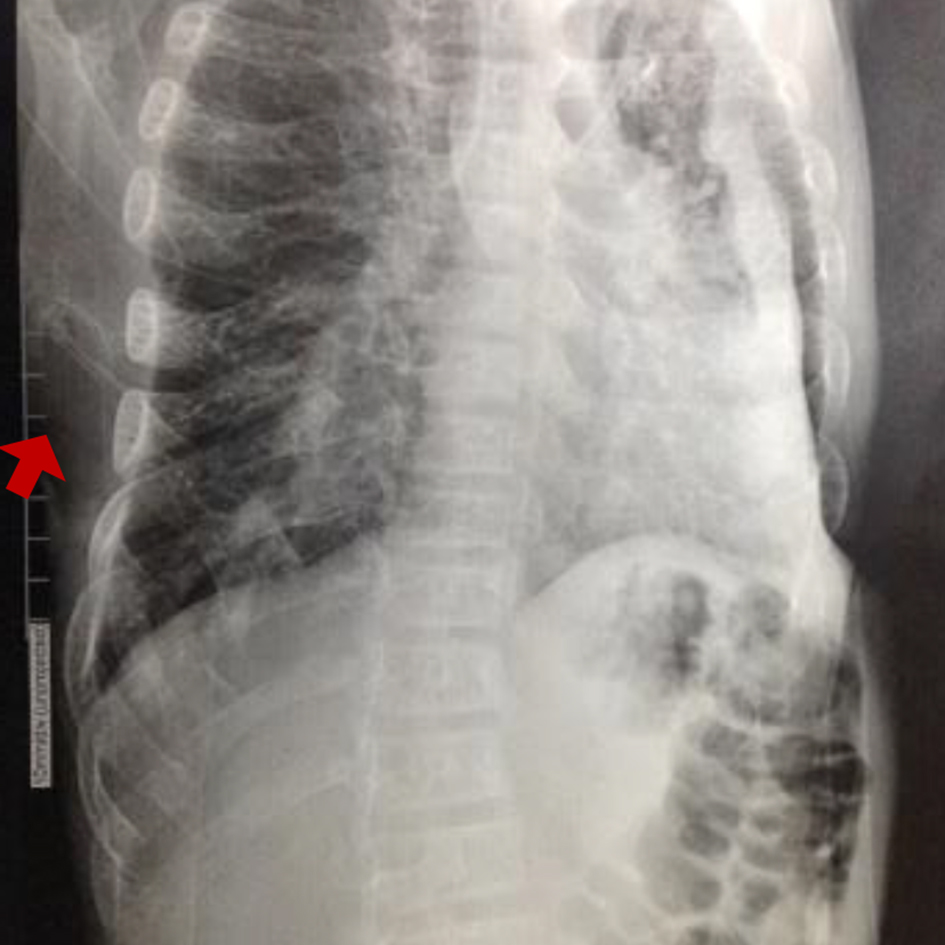 Click for large image | Figure 8. Narrow thorax, chest deformity: the ribs appear swollen; beading and cupping are seen in their anterior ends, hypomineralization and scoliosis (arrow). |
The physical examination showed a height of 121 cm (-3 SD), a weight of 20 kg (-3 SD) and a head circumference of 51 cm. The following signs were observed: dolichocephaly, oligodontia, pectus carinatum, genu valgus, bilateral incurvated of tibiae and fibula. Genetic study revealed a homozygous mutation c.892 G>A (p.Glu298lys).
Enzyme replacement therapy with asfotase alfa was started. In the first 6 months of treatment, the patient improved his gait, increased his muscular strength and managed long distance walks in addition to jumping on his lower limbs.
Polysomnographic study performed at 15 years of age showed a severe sleep apnea-hypopnea syndrome (apnea-hypopnea index (AHI): 12.6/h) associated with desaturation and snoring. New polysomnographic study at 19 years of age reported mild apnea-hypopnea syndrome (AHI: 11.7/h). Spirometry and volume flow curve performed at 19 years of age reported restrictive ventilatory alteration that did not respond to the bronchodilator.
Case 7
The proband was a 9-year-old female, who is the youngest of three children and was born after a term pregnancy of 39 weeks of consanguineous parents.
At the age of 8 months, the mother evidenced deformity in spine and thorax. She received a diagnosis of idiopathic scoliosis and initiated evaluation by orthopedics. At age of 2 years, she was evaluated by a geneticist that gave a diagnosis of metaphyseal dysplasia. An early loss at 2 years of deciduous teeth was reported, and at the age of 5, due to the progression of deformity, a corset orthosis was indicated. Surgical management was considered due to the progression of scoliosis and pain. No seizures, fractures were observed.
The physical exam showed a height of 120 cm (-3.25), a weight of 20 kg (-3.4) and a head circumference of 50 cm (-2.30), with normal teeth, rib depression in the left hemithorax, pectus carinatum, asymmetry of scapulae, severe scoliosis and left thoracic gib. Her blood calcium, phosphorus and PTH levels were normal. There were no abnormal levels of amino acids or mucopolysaccharides in her urine analysis, and the serum ALP activity was low 27.4 U/L (reference value: 156 - 359 U/L).
At 7 years of age, the right thoracic curve measured 30°, which progressed to 40° by the age of 9 (Fig. 9). Radiographs of long bones documented that the proximal and distal metaphysis were significantly widened, with hypomineralized regions and physeal widening.
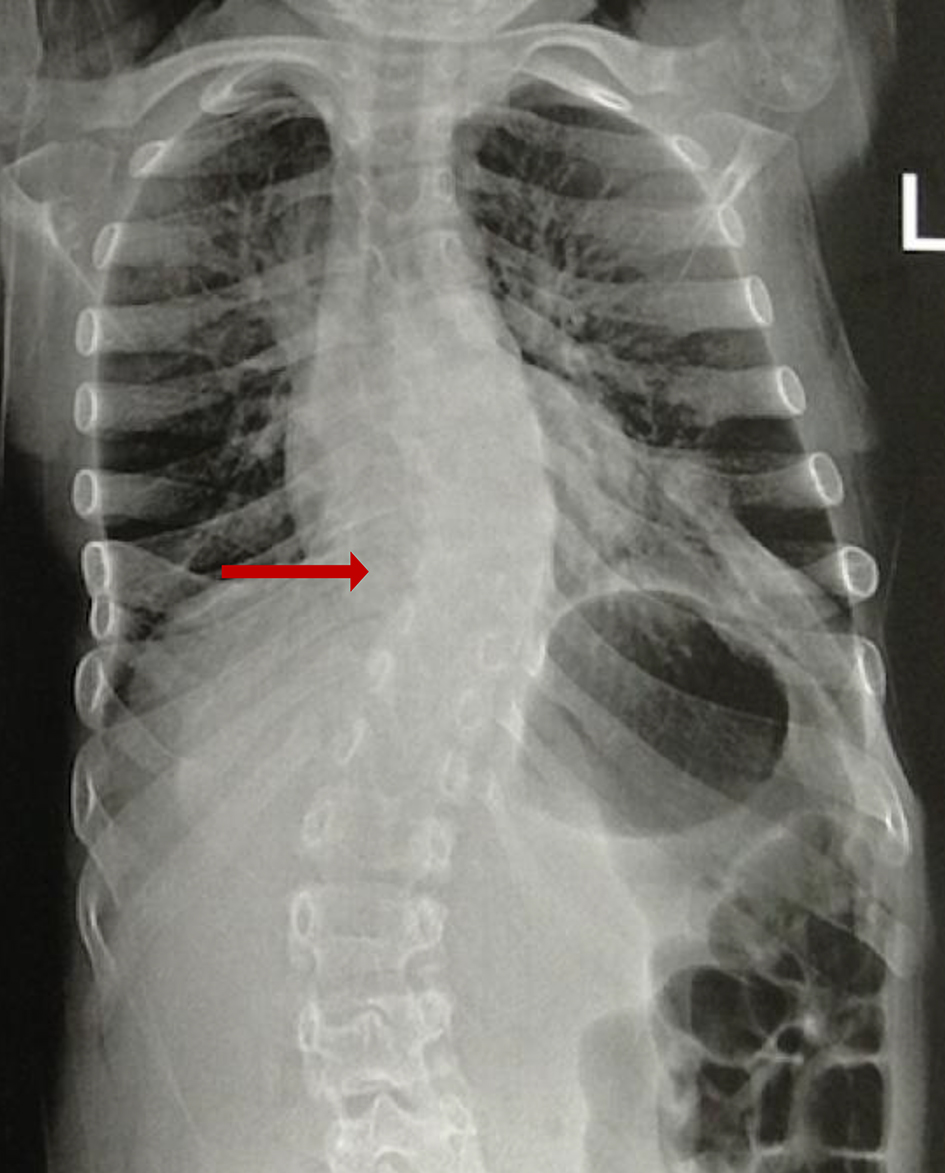 Click for large image | Figure 9. Spinal X-ray (2015): left scoliosis with a Cobb angle of 40° and decrease of bone mineralization (arrow). |
A homozygous c.892G>A (p.E298K) mutation was detected in the ALPL gene.
Her mother had a borderline serum ALP level of 44 U/L (reference value: 156 - 369 IU/L) and was heterozygous for the c.892G>A mutation. A bone mineral density scanning of the mother reported a normal z score and the levels in the proband’s sister was normal. They have no known family history of HPP.
Because she is having school issues, cognitive tests were conducted (WISC IV). They determined mild cognitive disability (CIT 63) in the patient.
After diagnosis, she started treatment with enzyme therapy, and surgical management of scoliosis was performed with adequate clinical and functional evolution. She had improvement in height, weight, pain in limbs, spine and more participation in physical activities.
Bone densitometry monitoring showed normal bone mineral density for the patient’s age, sex and race, and both improvement in bone mass and density.
| Discussion | ▴Top |
HPP was first described by Dr. John Campbell Rathbun in 1948 [7, 8]. It is a rare inherited metabolic disorder characterized by defective bone and teeth mineralization, and deficiency of serum and bone ALP activity caused by loss of function mutations within the gene that encodes the TNSALP. More than 411 mutations have been described and are responsible for the extraordinary clinical heterogeneity; added to the inheritance pattern, it can be autosomal recessive or dominant [5] (ALPL gene mutations database). Clinical presentation can be correlated with residual TNSALP activity and/or dominant-negative effects, and for this reason, this disease is considered a continuous spectrum. Six clinical forms are usually recognized based on age at diagnosis and severity of features. Perinatal form is seen in utero and is often lethal; however, there is a benign prenatal form when it shows a relatively benign postnatal course with spontaneous improvement of bowing with a less severe HPP phenotype [9]. As in case 1, he had an improvement of mineralization and good response to enzymatic therapy. Infantile HPP manifests before 6 months of age and patients appear normal until the onset of failure to thrive and rickets. The cranial sutures are wide, reflecting hypomineralization of the skull and rachitic deformity predisposes the infant to recurrent respiratory infections. The estimated mortality rate is around 50% without treatment [7]. Childhood form is commonly characterized by premature deciduous teeth loss, skeletal deformities, bone pain, short stature, delay in walking and/or impaired gait; history of fractures is frequent. The adult form usually presents in the middle age; the condition must be suspected in patients with recurrent metatarsal fractures, femoral pseudofractures, bone pain, pseudogout and chondrocalcinosis, and they can have history of premature loss of deciduous teeth [3, 10]. Odontohypophosphatasia is characterized by premature exfoliation of the primary teeth and severe dental caries. The anterior deciduous teeth are more likely to be affected; they do not have associated skeletal abnormalities [11, 12].
HPP is a panethnic and worldwide disease. In Canada, the estimated prevalence of the severe form is approximately 1/100,000 and is particularly high in the Mennonite Canadian population (1/2,500 and 1/25 might be carriers). In Europe, the prevalence of the severe form is 1/300,000 and 2 - 3/1,000,000 in Japan and rare in African-Americans [12]. Moderate forms of HPP are likely more frequent because heterozygotes may be affected. The prevalence of milder forms is difficult to estimate, being possibly as high as 1 in 6.370 [3, 5, 8].
We report seven patients with diagnosis of HPP with confirmed clinical, biochemical and molecular diagnosis. Four different mutations were identified, and all of them were missense variants. The most common form of HPP in patients was childhood HPP. All variants are included in ALPL gene mutations database (http://www.sesep.uvsq.fr/03_hypo_mutations.php), in whom five had the same mutation 892G>A (p.Glu298Lys) (four homozygous and one heterozygous state). They presented mostly in the childhood form. This mutation was reported by Orimo et al, in a compound heterozygous state, in a Japanese infant girl who exhibited profound skeletal hypomineralization and respiratory distress and died at age of 5 months and 27 days [13]. Case 1 was homozygous for this variant and in contrast with the other patients with the same genotype and who presented the juvenile form, he presented suggestive in utero imaging changes and significant compromise at birth and subsequent improvement in mineralization, for which he was classified as a benign perinatal form. This reinforces the variable expressivity described previously in this disease.
Cases 3 to 7 experienced a delay in diagnosis, although the onset of their signs and symptoms was evident from childhood and all patients had low serum ALP activity, and the clinical manifestations were very broad.
Case 4 was heterozygous for variant c.334G>C (p.Gly112Arg) and he had a childhood form. This variant had been previously reported by Whyte et al, in a study of 173-patient cohort, with the objective of evaluating the extant nosology for HPP in children. In contrast, patient had only dental manifestations and he was classified as odontohypophosphatasia [14]. So far, we have not known more cases reported in the literature to date for this variant.
Finally, case 2 was compound heterozygous for variants c.394G>A (p.Ala132Thr) and c.571G>A (p.Glu191Lys); both variants have been individually reported in the literature but have been associated with different phenotypes [15, 16].
The variant c.394G>A (p.Ala132Thr) has been related to patients having adult onset HPP and odontohypophosphatasia combined with other mutations. Variant c.571G>A (p.Glu191Lys) is a very common mutation reported in European population with moderate HPP, and it was recently reported in two Chinese patients with childhood HPP [17]. Tenorio et al reported two patients with this variant, one compound heterozygous p.(Glu191Lys); (Asp378Gly) from Portugal with infantile HPP and the other heterozygous for the variant from Russian with adult HPP [12].
Case 7 has associated mild cognitive disability, which has been observed in all types of HPP, including patients with or without complications from hypoxic event secondary to respiratory problems, and this finding should be taken into account when diagnosing and monitoring patients [18, 19].
Del Angel et al measured residual activity and dominant-negative effect (DNE) for 155 ALPL variants by transient transfection and TNSALP enzymatic activity measurement. In this study, two of the four mutations found in our population were evaluated. Variant p.Glu298Lys in exon 9 is located in a calcium binding site, and it has not a DNE. In vitro functional studies showed a relative value of TNSALP enzymatic activity in homozygous of 23.8% with respect to wild type (WT) [16].
Additionally, variant p.Glu191Lys in exon 6 is an unspecified location and it has not a DNE; the enzymatic activity in homozygous was 21.4% compared with 58.7% in heterozygous vs. WT [16].
In this article, we present different patients with HPP with the objective of contributing to the understanding of this rare disease and describing the Colombian population.
Learning points
It is important to highlight that despite the fact that the patients presented symptoms from an early age and they had low or limited values of low serum total ALP for age and gender, the vast majority were diagnosed several years later, and it can be due to limited awareness of HPP. The most frequent clinical manifestations were short stature and dental compromise.
The variant 892G>A (p.Glu298Lys) was the most frequent mutation in these populations in homozygous and heterozygous state, also presenting a high variability of phenotypes since perinatal benign until childhood form, which is associated with a wide spectrum in severity and presentation. It results in a difficulty for predicting the severity and giving a prognosis in patients with this variant or with novel mutations in ALPL gene. Finally, this suggests other factors that influence phenotype such as: environmental, epigenetic and modifier genes.
Currently, enzyme replacement therapy with asfotase alfa (Strensiq™) is the only approved treatment for HPP. Asfotase alfa can restore normal ALP levels and prevents or improves skeletal manifestations and respiratory complications of HPP. Five of the patients included in this report have received recombinant TNSALP replacement therapy, with an adequate safety and tolerance profile, as well as clinical, radiological improvement and an acceptable postoperative result.
Acknowledgments
The authors thank all the patients and their family members for their involvement in the present study.
Financial Disclosure
We are requesting a sponsorship of Alexion.
Conflict of Interest
The authors declare that they have no conflict of interest.
Informed Consent
The parent of each patient or patient provided written informed consent for publication of medical data and genetic data.
Author Contributions
This work was carried out in collaboration between all authors. AMZ and JCP wrote the manuscript. All authors read and approved the final manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article. Case 7 was reported previously, but in this paper included new data.
| References | ▴Top |
- Gorodischer R, Davidson RG, Mosovich LL, Yaffe SJ. Hypophosphatasia: a developmental anomaly of alkaline phosphatase? Pediatr Res. 1976;10(7):650-656.
doi pubmed - Rathbun JC. Hypophosphatasia; a new developmental anomaly. Am J Dis Child. 1948;75(6):822-831.
doi pubmed - Choida V, Bubbear JS. Update on the management of hypophosphatasia. Ther Adv Musculoskelet Dis. 2019;11:1759720X19863997.
doi pubmed - Kishnani PS, Rush ET, Arundel P, Bishop N, Dahir K, Fraser W, Harmatz P, et al. Monitoring guidance for patients with hypophosphatasia treated with asfotase alfa. Mol Genet Metab. 2017;122(1-2):4-17.
doi pubmed - Zarante Bahamon AM. Severe scoliosis in a Colombian patient with childhood hypophosphatasia. Clin Cases Miner Bone Metab. 2018;15:364-367.
- Szabo SM, Tomazos IC, Petryk A, Powell LC, Donato BMK, Zarate YA, et al. Frequency and age at occurrence of clinical manifestations of disease in patients with hypophosphatasia: a systematic literature review. Orphanet J Rare Dis. 2019;14(1):1-9.
doi pubmed - Chou YY, Ou HY, Wu TJ, Tsai SC, Lin SJ, Yu EH. Hypophosphatasia in Taiwan: report of two cases. Kaohsiung J Med Sci. 2005;21(3):134-137.
doi - Mumm S, Jones J, Finnegan P, Whyte MP. Hypophosphatasia: molecular diagnosis of Rathbun's original case. J Bone Miner Res. 2001;16(9):1724-1727.
doi pubmed - Matsushita M, Kitoh H, Michigami T, Tachikawa K, Ishiguro N. Benign prenatal hypophosphatasia: a treatable disease not to be missed. Pediatr Radiol. 2014;44(3):340-343.
doi pubmed - Mornet E, Nunes ME. Hypophosphatasia. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. Gene Reviews. Seattle (WA): University of Washington, Seattle; 2007.
- Mori M, DeArmey SL, Weber TJ, Kishnani PS. Case series: Odontohypophosphatasia or missed diagnosis of childhood/adult-onset hypophosphatasia? - Call for a long-term follow-up of premature loss of primary teeth. Bone Rep. 2016;5:228-232.
doi pubmed - Mao X, Liu S, Lin Y, Chen Z, Shao Y, Yu Q, Liu H, et al. Two novel mutations in the ALPL gene of unrelated Chinese children with Hypophosphatasia: case reports and literature review. BMC Pediatr. 2019;19(1):456.
doi pubmed - Mornet E, Yvard A, Taillandier A, Fauvert D, Simon-Bouy B. A molecular-based estimation of the prevalence of hypophosphatasia in the European population. Ann Hum Genet. 2011;75(3):439-445.
doi pubmed - Orimo H, Hayashi Z, Watanabe A, Hirayama T, Hirayama T, Shimada T. Novel missense and frameshift mutations in the tissue-nonspecific alkaline phosphatase gene in a Japanese patient with hypophosphatasia. Hum Mol Genet. 1994;3(9):1683-1684.
doi pubmed - Whyte MP, Zhang F, Wenkert D, McAlister WH, Mack KE, Benigno MC, Coburn SP, et al. Hypophosphatasia: validation and expansion of the clinical nosology for children from 25 years experience with 173 pediatric patients. Bone. 2015;75:229-239.
doi pubmed - Del Angel G, Reynders J, Negron C, Steinbrecher T, Mornet E. Large-scale in vitro functional testing and novel variant scoring via protein modeling provide insights into alkaline phosphatase activity in hypophosphatasia. Hum Mutat. 2020;41(7):1250-1262.
doi pubmed - Lefever E, Witters P, Gielen E, Vanclooster A, Meersseman W, Morava E, Cassiman D, et al. Hypophosphatasia in adults: clinical spectrum and its association with genetics and metabolic substrates. J Clin Densitom. 2020;23(3):340-348.
doi pubmed - Tenorio J, Alvarez I, Riancho-Zarrabeitia L, Martos-Moreno GA, Mandrile G, de la Flor Crespo M, Sukchev M, et al. Molecular and clinical analysis of ALPL in a cohort of patients with suspicion of Hypophosphatasia. Am J Med Genet A. 2017;173(3):601-610.
doi pubmed - Taketani T, Onigata K, Kobayashi H, Mushimoto Y, Fukuda S, Yamaguchi S. Clinical and genetic aspects of hypophosphatasia in Japanese patients. Arch Dis Child. 2014;99(3):211-215.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.