

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website https://www.jofem.org |
Case Report
Volume 10, Number 6, December 2020, pages 190-194
A Case of Etomidate Use in Management of Adrenocortical Carcinoma With Hypercortisolemia
Sharifah Faradila Wan Muhamad Hattaa, c, Rachel Dalyb, Cyril Chackob, Rajeev Raghavanb
aFaculty of Medicine, Universiti Teknologi MARA Sungai Buloh Campus, Jalan Hospital, 47000 Sungai Buloh, Selangor, Malaysia
bNew Cross Hospital, Royal Wolverhampton NHS Trust, Wolverhampton, UK
cCorresponding Author: Sharifah Faradila Wan Muhamad Hatta, Faculty of Medicine, Universiti Teknologi MARA Sungai Buloh Campus, Jalan Hospital, 47000 Sungai Buloh, Selangor, Malaysia
Manuscript submitted November 4, 2020, accepted November 9, 2020, published online December 22, 2020
Short title: Management of ACC With Etomidate
doi: https://doi.org/10.14740/jem682
| Abstract | ▴Top |
Adrenocortical carcinoma (ACC) is rare with an incidence of 0.7 - 2.0 per million per year. Approximately 60% of cases present with hypercortisolism, and rapidly progressing Cushing’s syndrome with or without virilisation is the most frequent presentation of ACC. Surgical intervention, aimed at removing the tumor and the source of cortisol or adrenocorticotropic hormone (ACTH), is the optimal treatment in most cases of Cushing’s syndrome. However, in hypercortisolemic states, surgical intervention has high rates of perioperative mortality and morbidity. Oral adrenal steroidogenesis inhibitors are commonly used, but in some cases they may not be tolerated or may not act quickly enough to bring down the cortisol level prior to surgery. Hence, occasionally, a more potent, immediate acting, parental agent, e.g., etomidate is necessary. We describe a case of ACC producing cortisol, which was complicated by an acute psychotic episode requiring intravenous etomidate for stabilization of the clinical condition prior to surgery.
Keywords: Adrenocortical carcinoma; Cushing’s syndrome; Hypercortisolemia; Young hypertension; Etomidate
| Introduction | ▴Top |
Adrenocortical carcinoma (ACC), a rare tumor has the propensity to produce and secrete steroids with the most frequent condition is a cortisol-secreting ACC causing Cushingoid phenotype. The goals of treatment in ACC include both control of tumor growth and mitigation of the effects derived from the production of hormone excess, as hypercortisolism may lead to poor quality of life and precocious death. Therefore, rapid control of hormone over-secretion is important. Medications available include metyrapone, ketoconazole and etomidate [1]. Both metyrapone and ketoconazole may take time to work and are often inadequate in the face of life-threatening complications and limited oral intake conditions.
Etomidate is the only intravenous medical therapy for hypercortisolemia [2]. It is useful for rapid normalization of cortisol, in patients with severe Cushing’s syndrome who require urgent control but are unable to tolerate oral medication, such as patients presenting with acute psychosis [3]. Previous case reports have found that intravenous infusion of etomidate has been useful for patients with adrenocorticotropic hormone (ACTH)-secreting lung tumors, presenting with acute psychosis, although similar reports for patients with glucocorticoid-secreting ACCs are limited [4, 5]. We report a case of a patient with a cortisol-producing ACC, causing acute psychosis and hypokalemia, which was successfully managed with intravenous etomidate, prior to adrenalectomy.
| Case Report | ▴Top |
A 23-year-old lady was admitted to the hospital with symptoms of generalized lethargy, weakness, rapid weight gain, increasing abdominal girth and recurrent genital thrush over the past 6 months. She had history of Perthes’ disease, bronchial asthma, primary hypothyroidism, recently diagnosed hypertension and pulmonary stenosis corrected by pulmonary valvuloplasty. She was on levothyroxine 50 µg/day and amlodipine 10 mg/day. She was a non-smoker and did not drink any alcohol. There was no significant family history.
Clinical examination revealed: pulse rate of 123 beats per minute, blood pressure 162/98 mm Hg and body mass index 27.2 kg/m2. She had typical features of Cushing’s syndrome including moon facies, central obesity, 1 cm wide purple abdominal striae, infra-scapular fat pad, proximal myopathy and abdominal distension. The 24-h urinary cortisol was 2,182 nmol/24 h (normal (N): 0 - 130), random cortisol 800 nmol/L and ACTH < 0.3 ng/L (N: 0 - 50 ng/L). She was hypokalemic and endocrine tests confirmed hyperandrogenemia and suppression of the gonadal axis (Table 1).
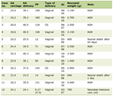 Click to view | Table 1. Blood Results on Presentation to Emergency Department |
Computed tomography of abdomen and chest showed a large multi-loculated, septate, cystic left adrenal mass measuring 15 × 10 × 8 cm. The mass was contiguous with the left renal vein giving a filling defect, suspicious of tumor thrombus. Bilateral lung metastases were noted. The conclusion was that the patient had a large lobulated adrenal carcinoma with involvement of the left renal vein and bilateral widespread metastatic lung lesions (Figs. 1, 2), American Joint Committee on Cancer (AJCC) and European Network for the Study of Adrenal Tumors (ENSAT) stage IV.
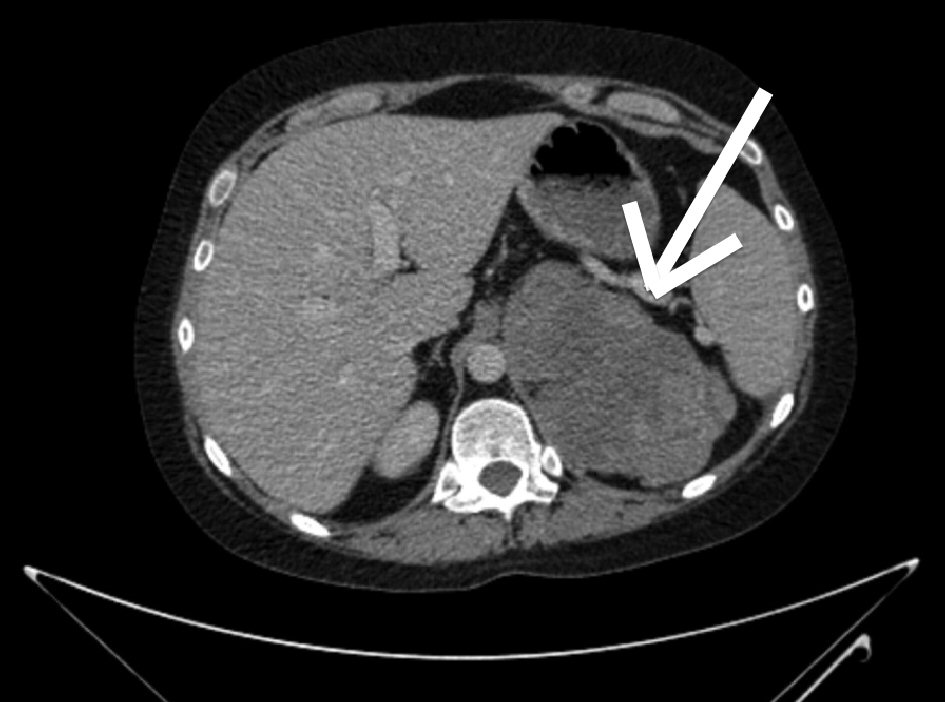 Click for large image | Figure 1. Computed tomography of the abdomen revealed a large multi-loculated, septate, cystic left adrenal mass measuring 15 × 10 × 8 cm. |
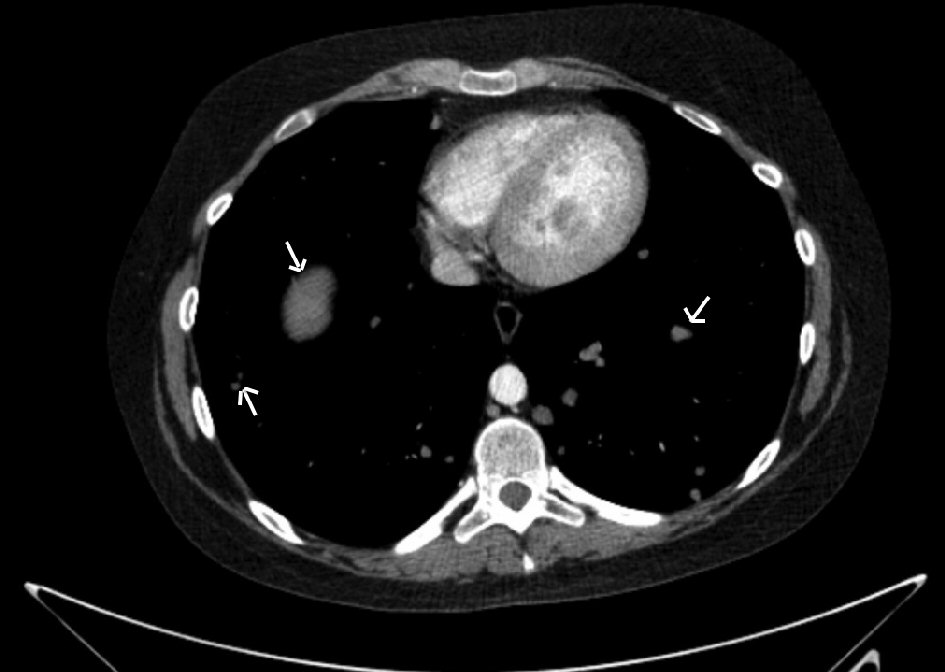 Click for large image | Figure 2. Computed tomography of the thorax revealed multiple lung metastases. |
While she was being investigated on the endocrine ward, on day 3 of admission, she developed fever, cough productive of sputum, and complained of difficulty in breathing at rest. In addition, she also became acutely confused, agitated and verbally aggressive; and it became progressively more difficult to manage her on the endocrine ward. Chest X-ray confirmed the presence of pneumonia with consolidation on the right lower zone. She was transferred to the intensive care unit (ICU) where she was intubated, ventilated and commenced on broad-spectrum intravenous antibiotics for hospital-acquired pneumonia, and intravenous etomidate 42.86 µg/kg/h for acute control of hypercortisolism. She was also started on ketoconazole 400 mg three times daily along with 1 mg dexamethasone as replacement corticosteroid. Cortisol level was monitored every 12 h aiming for a level between 400 - 500 nmol/L. Sequential cortisol levels and the dose of etomidate are depicted in Table 2.
Click to view | Table 2. Dose of Etomidate and Cortisol During the ICU Admission |
Following the initiation of etomidate infusion, cortisol level came down progressively and hemodynamic and metabolic parameters normalized. However it also resulted in gradual drop of hemoglobin level by 4 g with some of the hematological parameters suggestive of hemolysis (Table 3). She received blood transfusion and the dose of etomidate was gradually reduced and eventually discontinued on day 4 on ICU. Cortisol level at the time of withdrawal of etomidate was 210 nmol/L. At this stage unfortunately she developed ventilator-acquired pneumonia, which delayed extubation for 2 more days whilst she was successfully managed with an upgrade of broad-spectrum antibiotics. Following extubation, she remained stable and was transferred initially to the endocrine ward and then to the regional referral center.
Click to view | Table 3. Blood Results From Day 3 of ICU Admission |
Throughout the admission, her blood glucose remained within normal limits of between 6 - 10 mmol/L not requiring any form of oral anti-diabetic medication nor insulin administration. Meanwhile her blood pressure was well controlled on amlodipine 10 mg once daily, doxazosin 2 mg once daily and bisoprolol 10 mg once daily. Meanwhile, the only biochemical derangement was her potassium, which was normalized with an addition of slow release potassium tablets of 1.2 g three times a day.
Following a multidisciplinary discussion in the regional referral center, she underwent left adrenalectomy and post operatively, she was initiated on mitotane and ketoconazole was replaced by metyrapone. Unfortunately her general health deteriorated and she passed away peacefully in the hospital 3 months after surgery.
| Discussion | ▴Top |
ACC is rare with an incidence of 0.7 - 2.0 per million per year. It more commonly affects young adults and those between the fourth and fifth decades of life. More women are diagnosed with ACC than men. Most ACCs develop sporadically but can also be associated with genetic conditions, such as Beckwith-Wiedemann syndrome, multiple endocrine neoplasia (MEN) 1, congenital adrenal hyperplasia, familial polyposis coli and B-catenin mutations [6]. Approximately 60% of the cases present with hypercortisolism and rapidly progressing Cushing’s syndrome with or without virilisation is the most frequent presentation of ACC [6]. Another common endocrine abnormality is hyperandrogenism which usually occurs in 1.4-15% patients [7]. Furthermore, in a patient with an adrenal mass, a high dehydroepiandrosterone sulphate (DHEA-S) concentration may suggest ACC whilst a low level is indicative of a benign adenoma [8].
Patients with an ACC producing cortisol can present with typical features of Cushing’s syndrome as was seen in our patient. In most patients with Cushing’s syndrome caused by benign causes hypercortisolemia progresses in an insidious manner unlike in ACC where it often develops rapidly and this can modify its presenting features with more prominent neurological and metabolic effects. Another factor that governs the presentation is the activation of the mineralocorticoid receptors by the excessively high level of cortisol leading to more significant hypertension and hypokalemia. This is compounded by the insufficient renal cortisol inactivation by 11β-hydroxysteroid dehydrogenase type 2 [6]. ACC can also be hormonally inactive and such patients would usually present with abdominal discomfort or fullness, nausea, vomiting or back pain caused by the mass effect of the large tumor [9].
Severe Cushing’s syndrome is defined as serum cortisol > 1,000 nmol/L, 24-h urinary free cortisol > four times the upper limit of normal and/or hypokalemia < 3.0 mmol/L with the development of at least one of the critical features including hypertension, hyperglycemia, ketoacidosis, gastrointestinal hemorrhage, heart failure, thromboembolism, progressive myopathy and acute psychosis. Severe Cushing’s syndrome is an acute emergency and is managed by correcting metabolic derangements and rapidly lowering the raised cortisol [3]. The medical therapy of hypercortisolemia is predominantly based on the inhibition of adrenal steroidogenesis at one or more enzymatic sites or alternatively by antagonism of the glucocorticoid receptor or the suppression of ACTH. Most commonly this is done via the oral route with metyrapone and/or ketoconazole but in certain cases, intravenous etomidate may be required to control the acute manifestations of severe hypercortisolism. Other available medications include glucocorticoid receptor antagonist mifepristone [2].
Etomidate is a general anesthetic agent, of the imidazole group. It is known to have an inhibitory effect on adrenal steroid synthesis, which outlasts its hypnotic effect, which was its first indication when it was synthesized in 1964 [10]. The steroidogenesis inhibition is the basis of its use in the treatment of severe Cushing’s syndrome, as it is currently the only intravenous medication available [11]. It is highly plasma bound and is metabolized to inactive metabolites by hepatic and plasma esterases. It is excreted in the urine and to a lesser degree in bile.
In our case, the patient had severe Cushing’s syndrome, as her serum cortisol was > 1,000 nmol/L, she was hypokalemic and had glucocorticoid-induced acute psychosis, requiring urgent treatment. As a result of the psychotic effect, she could not be safely and effectively managed, and a decision was made to commence intravenous infusion of etomidate in the ICU. Ketoconazole was given in conjunction with etomidate. The serum cortisol level fell from 1,041 to 107 within 48 h, as shown in Table 2. After 83 h, etomidate was discontinued, and the patient was eventually extubated after being treated for a ventilator-acquired pneumonia. The published recommendations for the management of ACTH-dependent Cushing’s syndrome in 2008, state that where oral treatment is not possible to ensure safe and rapid reduction of cortisol level, intravenous etomidate should be used especially in cases with Cushing’s-induced psychosis with severe metabolic disturbances [11]. It should be administered at a rate of 40 - 50 µg/kg/h, which adequately inhibits cortisol production but does not sedate, and the dose can be titrated with serum cortisol level which is in line with how we managed our patient [2]. Higher dose (i.e., > 50 µg/kg/h) is sometimes required during infusion due to an “escape” effect, caused by an increase in endogenous ACTH and cortisol secretion. Nonetheless, an increase in etomidate should block the higher concentration at the expense of induction of sedation [12]. When patients are rendered hypoadrenalism, demonstrated by serum cortisol levels of < 100 nmol/L, glucocorticoid replacement therapy should be initiated by means of either oral or intravenous hydrocortisone aiming at a cortisol level of around 500 - 800 nmol/L, as this is considered appropriate as a “physiological stress cortisol level” in an ICU setting [13]. In our case dexamethasone was used instead of hydrocortisone, as it does not affect the assay, hence giving a true reflection of endogenous cortisol level. It is of utmost importance that etomidate is given in an environment where close clinical, hemodynamic and biochemical monitoring is available such as a high dependency ward, with medical staff equipped with the capacity to support the airway if sedation does occur and to rapidly respond to hypocortisolism resulting from medical management.
Although there are many case reports in the literature discussing the use of etomidate for the treatment of Cushing’s syndrome, few discuss its use for acute psychosis due to a glucocorticoid-secreting adrenal carcinoma. Bilgin et al and Baba et al discuss patients with ACTH-secreting lung tumors presenting with acute psychosis, which was successfully managed with intravenous etomidate [4, 5]. Wu et al and Tang et al both discuss patients with ACTH-independent adrenal tumors presenting with acute psychosis but treated with psychotropic medications prior to surgical resection. One of the patients discussed in Tang et al was also managed with metyrapone and mitotane preoperatively. All three patients discussed in the articles by Tang et al and Wu et al saw their psychotic symptoms gradually resolve postoperatively, and their psychotropic medications were discontinued. Both groups of authors discuss how psychotic presentation of Cushing’s syndrome can be easily overlooked and can lead to an increase in morbidity and mortality for patients if undiagnosed and untreated. It also emphasizes the importance of assessment for features of Cushingoid syndrome in patients presenting with acute psychosis and considering further biochemical and radiological investigations if Cushing’s syndrome is suspected [14, 15].
Common side effects of etomidate are nausea and vomiting, thrombophlebitis, pain on injection and myoclonic jerks during induction [10]. Central venous access is most appropriate when using the propylene glycol preparation. Safety measures should be undertaken by reducing the dose especially in patients who are elderly, very ill or have renal failure due to reduced protein binding and renal clearance [10]. Hemolysis has been suggested to be a side effect of the propylene glycol present in the preparation of etomidate [2]. The World Health Organization (WHO) suggests a maximum dose of 25 mg/kg per day of propylene glycol to minimize its toxic effects [2]. In our case, a diagnosis of hemolysis was suspected during the patient’s ICU admission, due to declining hemoglobin, raised reticulocytes and raised lactate dehydrogenase (LDH). However, the diagnosis could not be made unequivocally in view of normal haptoglobin and negative direct Coombs test (Table 3).
In conclusion, we present a case of ACC who presented with severe hypercortisolemia resulting in metabolic derangement and acute psychosis, and in whom the use of intravenous etomidate led to an excellent clinical and biochemical response. Patient safety was ensured during its administration for this less well-known indication by managing the patient in an intensive care setting where the patient received close monitoring.
Acknowledgments
None to declare.
Financial Disclosure
This research received no specific grant from any funding agency in the public, commercial, or not-for profit sectors.
Conflict of Interest
The authors declare that there is no conflict of interest.
Informed Consent
Written informed consent was obtained from the patient for the anonymized information to be published in this article.
Author Contributions
Sharifah Faradila Wan Muhamad Hatta: writing, editing, case detection, investigation and medical management; Rachel Daly: writing; Cyril Chacko: medical management in the Intensive Care Unit; Rajeev Raghavan: medical management in the medical ward.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Veytsman I, Nieman L, Fojo T. Management of endocrine manifestations and the use of mitotane as a chemotherapeutic agent for adrenocortical carcinoma. J Clin Oncol. 2009;27(27):4619-4629.
doi pubmed - Preda VA, Sen J, Karavitaki N, Grossman AB. Etomidate in the management of hypercortisolaemia in Cushing's syndrome: a review. Eur J Endocrinol. 2012;167(2):137-143.
doi pubmed - Alexandraki KI, Grossman AB. Therapeutic strategies for the treatment of severe Cushing's syndrome. Drugs. 2016;76(4):447-458.
doi pubmed - Baba M, Ray D. Severe psychosis due to Cushing's syndrome in a patient with a carcinoid tumour in the lung: a case report and review of the current management. World J Surg Oncol. 2015;13:165.
doi pubmed - Bilgin YM, van der Wiel HE, Fischer HR, De Herder WW. Treatment of severe psychosis due to ectopic Cushing's syndrome. J Endocrinol Invest. 2007;30(9):776-779.
doi pubmed - Else T, Kim AC, Sabolch A, Raymond VM, Kandathil A, Caoili EM, Jolly S, et al. Adrenocortical carcinoma. Endocr Rev. 2014;35(2):282-326.
doi pubmed - Dobbie JW. Adrenocortical nodular hyperplasia: the ageing adrenal. J Pathol. 1969;99(1):1-18.
doi pubmed - Fassnacht M, Kenn W, Allolio B. Adrenal tumors: how to establish malignancy ? J Endocrinol Invest. 2004;27(4):387-399.
doi pubmed - Allolio B, Fassnacht M. Clinical review: Adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab. 2006;91(6):2027-2037.
doi pubmed - Forman SA. Clinical and molecular pharmacology of etomidate. Anesthesiology. 2011;114(3):695-707.
doi pubmed - Biller BM, Grossman AB, Stewart PM, Melmed S, Bertagna X, Bertherat J, Buchfelder M, et al. Treatment of adrenocorticotropin-dependent Cushing's syndrome: a consensus statement. J Clin Endocrinol Metab. 2008;93(7):2454-2462.
doi pubmed - Mullan KR, Atkinson AB. Endocrine clinical update: where are we in the therapeutic management of pituitary-dependent hypercortisolism? Clin Endocrinol (Oxf). 2008;68(3):327-337.
doi pubmed - Heyn J, Geiger C, Hinske CL, Briegel J, Weis F. Medical suppression of hypercortisolemia in Cushing's syndrome with particular consideration of etomidate. Pituitary. 2012;15(2):117-125.
doi pubmed - Wu Y, Chen J, Ma Y, Chen Z. Case report of Cushing's syndrome with an acute psychotic presentation. Shanghai Arch Psychiatry. 2016;28(3):169-172.
- Tang A, O'Sullivan AJ, Diamond T, Gerard A, Campbell P. Psychiatric symptoms as a clinical presentation of Cushing's syndrome. Ann Gen Psychiatry. 2013;12(1):23.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.