

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website http://www.jofem.org |
Case Report
Volume 8, Number 6, December 2018, pages 149-152
A Case of Mitochondrial Disease First Diagnosed Over 30 Years After Onset
Ryo Watanabea, Hidekatsu Yanaia, c, Haruka Kamataa, Hiroko Yamazakib
aDepartment of Internal Medicine, National Center for Global Health and Medicine Kohnodai Hospital, Chiba, Japan
bDepartment of Ophthalmology, National Center for Global Health and Medicine Kohnodai Hospital, Chiba, Japan
cCorresponding Author: Hidekatsu Yanai, Department of Internal Medicine, National Center for Global Health and Medicine Kohnodai Hospital, 1-7-1 Kohnodai, Ichikawa, Chiba 272-8516, Japan
Manuscript submitted December 6, 2018, accepted December 21, 2018
Short title: Mitochondrial Disease Diagnosed Long After Onset
doi: https://doi.org/10.14740/jem542
| Abstract | ▴Top |
Mitochondrial disease is induced by mutations in both mitochondrial DNA and DNA of nuclear genes that encode mitochondrial proteins, and there is a general lack of genotype-phenotype correlation in many mitochondrial disorders, which makes the diagnosis of mitochondrial disease difficult. We treated a 37-year-old woman with congenital cataract, bilateral sensory deafness, severe bilateral ptosis, oculomotor disorder in all directions and systemic muscular atrophy and weakness. These clinical characteristics made us perform further examinations, and we found ischemic change in brain and remarkable atrophy of extraocular muscles by magnetic resonance imaging (MRI), optic atrophy by fundus examination and high levels of serum lactate and lactate/pyruvate ratio, which confirmed the diagnosis of mitochondrial disease. Finally, she was diagnosed with having mitochondrial disease for over 30 years after onset.
Keywords: Lactate; Mitochondrial disease; Ptosis; Pyruvate
| Introduction | ▴Top |
Mitochondrial disease has a complicated genetic property because two distinct genomes, mitochondrial DNA and DNA of nuclear genes that encode mitochondrial proteins, can contribute to disease pathogenesis [1, 2]. Therefore, mitochondrial dysfunction can arise because of defects in either mitochondrial DNA or nuclear mitochondrial genes, and can present in childhood or adulthood in association with vast clinical heterogeneity, with symptoms affecting a single organ or tissue, or multisystem involvement [2]. There is no effective treatment for mitochondrial disease. However, we think that if we diagnose the mitochondrial disease early, we can improve patients’ wellbeing and alleviate misfortune. Here, we describe a case of mitochondrial disease that took more than 30 years to diagnose to raise attention for mitochondrial disease in the medical community.
| Case Report | ▴Top |
A 37-year-old woman was emergently admitted to our hospital due to severe leanness, isolated from parents and accompanied by city official staffs. She was diagnosed with congenital cataract and bilateral sensory deafness at the age of 0 and 3 years, respectively. At the young age, she often shouted and was panic. She graduated from high school and entered caregiver vocational school. However, she could not continue to practice due to severe muscle weakness.
On admission, her height, weight and body mass index were 147.9 cm, 29.7 kg and 13.6 kg/m2, respectively with clear consciousness. She showed vital signs as follows: body temperature, 37.0 °C; heart rate, 90 beats per minute (bpm); blood pressure, 119/75 mm Hg, and respiratory rate, 18/min. Bilateral severe ptosis and oculomotor disorder in all directions were observed (Fig. 1). Systemic muscular atrophy and weakness were also observed.
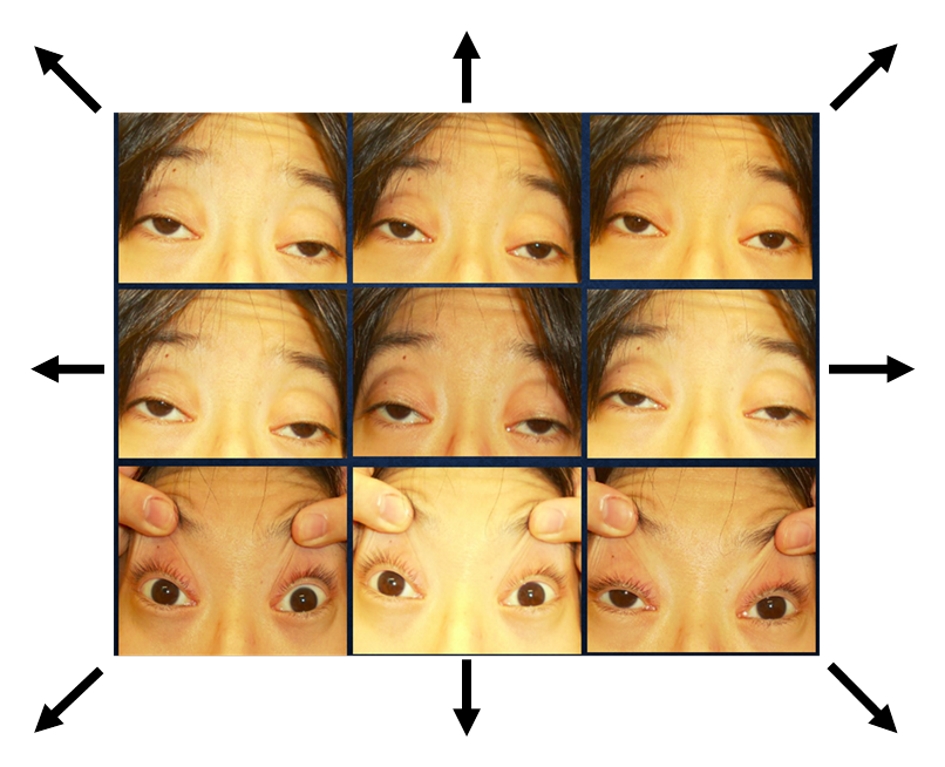 Click for large image | Figure 1. Bilateral severe ptosis and oculomotor disorder in all directions were observed. Arrows show the direction indicated to see. |
Laboratory data on admission were shown in Table 1. Serum levels of creatine kinase, aspartate transaminase and lactate dehydrogenase were elevated, reflecting muscular weakness. Serum creatinine level was very low, indicating severe muscular atrophy.
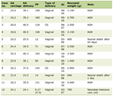 Click to view | Table 1. Laboratory Data on Admission |
The fundus examination of both eyes showed pale optic disc and narrowing of vessels, indicating the existence of optic atrophy (Fig. 2). Brain T2-weighted magnetic resonance imaging (MRI) showed diffuse high-intensity area (Fig. 3a), resembling ischemic change. Remarkable atrophy of extraocular muscles in bilateral eyes was also observed (Fig. 3b).
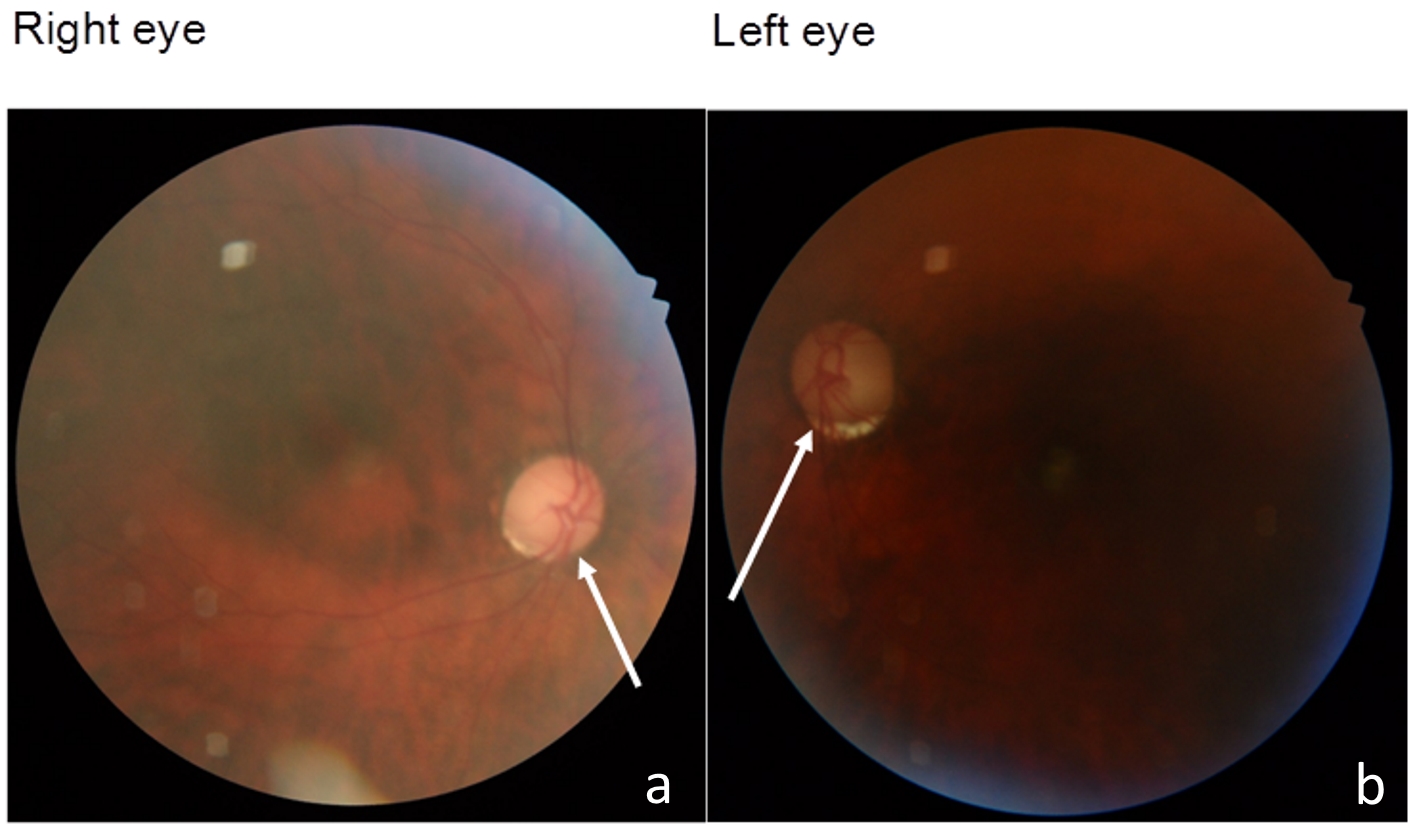 Click for large image | Figure 2. The fundus examination of both eyes showed pale optic disc (arrows) and narrowing of vessels. |
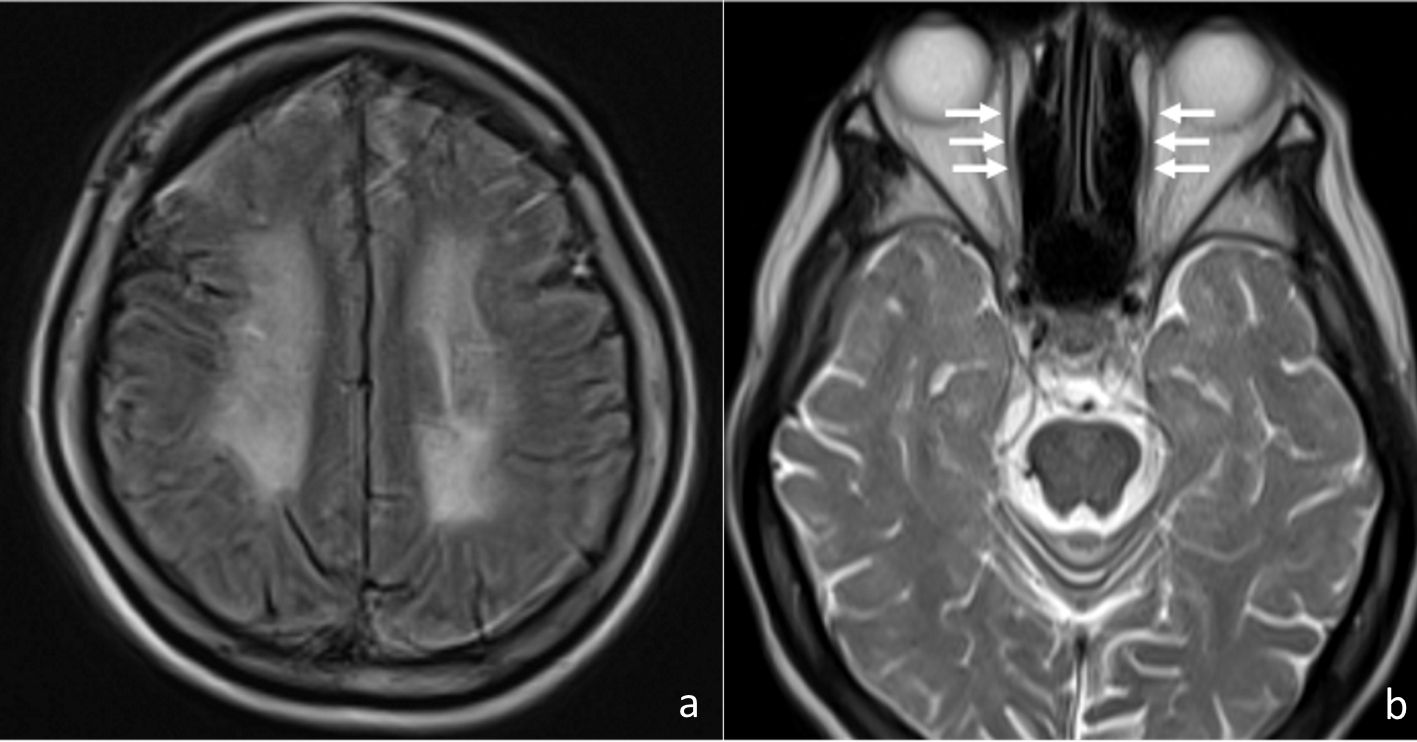 Click for large image | Figure 3. Brain T2-weighted MRI showed diffuse high-intensity area (a), and remarkable atrophy of extraocular muscles (arrows) in bilateral eyes were observed (b). |
After these examinations, she was suspected of having mitochondrial disease, and we measured the serum lactate and pyruvate levels. Serum lactate and pyruvate levels were 19.0 mg/dL (normal range, 4.2 - 17.0 mg/dL) and 0.59 mg/dL (normal range, 0.3 - 0.9 mg/dL), and the lactate/pyruvate ratio was 32 (normal range, < 10). Serum lactate level was also high (24.6 mg/dL) on another day. Finally, she was diagnosed with having mitochondrial disease. However, serum coenzyme Q10 level (517 ng/mL) had been kept within normal reference range (338 - 1340 ng/mL). Further, commercially measurable point mutations such as A1555G, T3271C, A8344G, G8363A, T8993G/C, T9176C, G11778A, G13513A, and A3243G were not detected.
Nutritional and physical therapy slightly improved her physical ability, but administration of coenzyme Q10 did not influence her symptoms. However, she was able to apply for the Ministry of Health Research Project for the Treatment of Special Chronic Diseases, and could obtain medical and social support.
Before submitting this article, we obtained written permission to report from this patient.
| Discussion | ▴Top |
Mitochondria have a very crucial role in cellular bioenergetics and cell death, and are important to support cellular integrity such as function and death process. The mitochondrial respiratory chain deficiency has been suggested to induce the pathogenesis of a wide range of neurological disorders [3, 4]. Mitochondrial Ca2+ accumulation is a tightly controlled process, in turn regulating functions as diverse as aerobic metabolism and induction of cell death [3]. Mitochondria are critical regulators of cell death, a key feature of neurodegeneration. Mutations in mitochondrial DNA and oxidative stress both contribute to the development of neurodegenerative diseases [4].
Therefore, neurological symptoms are particularly common, and may be devastating in patients with mitochondrial disease, including sensorineural deafness, cerebellar ataxia, peripheral neuropathy, dementia, and epilepsy [5]. Various eye signs accompany mitochondrial disease including ptosis, progressive external ophthalmoplegia, optic atrophy, retinopathy, and cataract [5, 6]. Ptosis, external ophthalmoplegia, optic atrophy and cataract which were observed in our patient helped us diagnose mitochondrial disease. Optic neuropathy is very difficult to treat. The treating ophthalmologist should maximize any residual visual acuity by the use of visual aids and should help the patient to gain access to financial and other social support by formally registering the patient as blind [5], which was provided to our patient. Corrective ptosis surgery can dramatically improve both visions. However, given the progressive nature of the muscle disease, a detailed preoperative assessment of eye movements is essential [5]. We thought that she could not obtain the improvement for her visual acuity by operation; therefore, she did not undergo operation.
Treatment of mitochondrial disease is extremely difficult, and coenzyme Q10 is the only agent which has shown some therapeutic benefit for the patients [7]. Coenzyme Q10 serves as an electron carrier in the mitochondrial respiratory chain as well as functioning as a potent lipid soluble antioxidant [7]. However, we could not observe any beneficial effects of coenzyme Q10 on our patient’s symptoms of mitochondrial disease. Coenzyme Q10 can be effective for early mitochondrial disease, but may not be effective for late-phase mitochondrial disease.
The therapies targeting basic mitochondrial processes, such as energy metabolism or free-radical generation, or specific interactions of disease-related proteins with mitochondria, hold great promise [4]. However, in any case, early diagnosis of mitochondrial disease may be important to receive promising therapy.
There is a general lack of genotype-phenotype correlation in many mitochondrial disorders. Further, there is no definitive genetic diagnosis of mitochondrial disease. The measurement of serum lactate and pyruvate was helpful for us to diagnose mitochondrial disease. Especially, high lactate/pyruvate ratio was very supportive to diagnose mitochondrial disease. The analysis of serum biomarkers and magnetic resonance spectroscopy (MRS) in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) showed that the severity of serum lactate/pyruvate was correlated with severity of the MRS lactate peak (P = 0.0007) in regions with acute infarcts, and the severity of the MRS lactate peak was also weakly correlated with severity of serum lactate/pyruvate (P = 0.03) in previously infarcted regions [8], supporting the usefulness of serum lactate/pyruvate ratio for diagnosis of mitochondrial disease. Sasaki et al reported that lactate/pyruvate ratio tended to be higher in patients with mitochondrial respiratory chain disorder than those without the disorder [9]. From the receiver operating characteristic curve, the optimal cutoff value of lactate/pyruvate was 23.2, suggesting lactate/pyruvate ratio can be excellent predictors of mitochondrial respiratory chain disorder.
Short stature, ptosis, external ophthalmoplegia, cataract and sensory deafness made us think of mitochondrial disease. These clinical characteristics led us to perform further examinations such as MRI, fundus examination of eyes and measurement of serum lactate and pyruvate, which confirmed the diagnosis of mitochondrial disease.
Conclusions
We experienced a case of mitochondrial disease first diagnosed at over 30 years after onset.
Competing Interest
The authors declare that they have no competing interest concerning this article.
| References | ▴Top |
- Craven L, Alston CL, Taylor RW, Turnbull DM. Recent advances in mitochondrial disease. Annu Rev Genomics Hum Genet. 2017;18:257-275.
doi pubmed - Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW. The genetics and pathology of mitochondrial disease. J Pathol. 2017;241(2):236-250.
doi pubmed - Celsi F, Pizzo P, Brini M, Leo S, Fotino C, Pinton P, Rizzuto R. Mitochondria, calcium and cell death: a deadly triad in neurodegeneration. Biochim Biophys Acta. 2009;1787(5):335-344.
doi pubmed - Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787-795.
doi pubmed - McFarland R, Taylor RW, Turnbull DM. A neurological perspective on mitochondrial disease. Lancet Neurol. 2010;9(8):829-840.
doi - Finsterer J, Zarrouk-Mahjoub S, Daruich A. The Eye on Mitochondrial Disorders. J Child Neurol. 2016;31(5):652-662.
doi pubmed - Hargreaves IP. Coenzyme Q10 as a therapy for mitochondrial disease. Int J Biochem Cell Biol. 2014;49:105-111.
doi pubmed - Lee HN, Yoon CS, Lee YM. Correlation of serum biomarkers and magnetic resonance spectroscopy in monitoring disease progression in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes due to mtDNA A3243G mutation. Front Neurol. 2018;9:621.
doi pubmed - Sasaki K, Sakamoto S, Uchida H, Narumoto S, Shigeta T, Fukuda A, Ito R, et al. liver transplantation for mitochondrial respiratory chain disorder: a single-center experience and excellent marker of differential diagnosis. Transplant Proc. 2017;49(5):1097-1102.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.