

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website http://www.jofem.org |
Original Article
Volume 7, Number 2, April 2017, pages 55-60
Calcium Mediates the Phosphaturia in Hypercalcemia of Malignancy
Issac Sachmechia, c, Veronica Jedlovskya, George Schusslerb
aDivision of Endocrinology and Metabolism, Icahn School of Medicine at Mount Sinai Services-Queens Hospital Center, Jamaica, NY, USA
bDivision of Endocrinology and Metabolism, State University Hospital Downstate, Brooklyn, NY, USA
cCorresponding Author: Issac Sachmechi, Division of Endocrinology and Metabolism, Department of Medicine, Icahn School of Medicine at Mount Sinai Services-Queens Hospital Center, 82-68 164th Street, Jamaica, NY 11432, USA
Manuscript accepted for publication March 29, 2017
Short title: Phosphaturia in Hypercalcemia of Malignancy
doi: https://doi.org/10.14740/jem402w
| Abstract | ▴Top |
Background: Humoral hypercalcemia of malignancy (HHM) is claimed to be caused by circulating parathyroid hormone like peptide (PTHrP) that shares several of the cardinal biochemical and skeletal histological features with hyperparathyroidism (HPT). Both syndromes are characterized by hypercalcemia from increased renal tubular reabsorption of filtered calcium, increased urinary cyclic AMP, renal phosphate wasting and hypophosphatemia, in contrast to low 1,25(OH)2D levels, metabolic alkalosis, and uncoupled bone formation-resorption rate in HHM. The aim of the study was to reassess if phosphaturia of HHM can be attributed to hypercalcemia rather than to humoral effect of PTHrP.
Methods: A prospective study was conducted at Mount Sinai Services at Queens Hospital Center, NY.
Results: Nine (four females and five males) out of 20 patients with hypercalcemia of malignancy documented having elevated PTHrP, normal renal function, and low PTH levels were finalized for the study. Serum calcium, phosphate, creatinine, albumin, and PTHrP concentration were determined. On the initiation of hydration, 24 h urine for creatinine, calcium and phosphorous were also collected. Blood and urine tests were repeated after serum calcium levels had been decreased by at least 1.5 mg/dL. All patients received vigorous intravenous hydration with normal saline at 200 cc/h. After the first 24 h, small doses of furosemide were given to two out of nine patients for 48 - 72 h. Hydration has shown statistically significant decrease in serum calcium concentration (13.9 ± 2.1 to 10.8 ± 1.48 mg/dL; P < 0.001) and increase in tubular reabsorption of phosphate (TRP) (69.54±8.16% to 77.85±12.24%; P < 0.01). However, PTHrP levels have shown no statistically significant difference on hydration therapy.
Conclusion: Improvement of hypercalcemia by hydration reverses phosphaturia without reducing PTHrP levels. It is being proposed that in HHM, PTHrP can lead to hypercalcemia from uncoupled bone formation-resorption rate through a local autocrine or paracrine action produced by bony micrometastases that could not be demonstrated radiologically, rather than from PTHrP’s humoral effect.
Keywords: Hypercalcemia of malignancy; Parathyroid hormone like peptide; Phosphaturia; Tubular reabsorption of phosphate
| Introduction | ▴Top |
Humoral hypercalcemia of malignancy (HHM) and hyperparathyroidism (HPT) share several of the cardinal biochemical and skeletal histological features. Both syndromes are characterized by hypercalcemia from increased renal tubular reabsorption of filtered calcium, increased urinary cyclic AMP (cAMP), renal phosphate wasting and hypophosphatemia. In contrast to these similarities, low 1,25(OH)2D3 concentrations, uncoupled bone formation and resorption, and metabolic alkalosis are observed in HHM which seems to be inconsistent with a humoral activation of the PTH receptor [1, 2].
Decreased renal tubular reabsorption of phosphate (TRP) and hypophosphatemia, commonly occurring in the HHM, have been attributed to a humoral effect of parathyroid hormone like peptide (PTHrP) which binds to and activates the PTH receptor [1]. An alternative explanation for the decreased TRP observed in HHM is hypercalcemia itself rather than PTHrP [3]. In fact, it has been shown that lowering serum calcium independently of any anticancer treatment reverses the decreased TRP as would be consistent with a phosphaturia effect of hypercalcemia rather than a humoral effect mediated by PTHrP [4]. However, these observations, made prior to the identification of PTHrP, did not rule out the possibility that the observed increase in TRP with treatment of the hypercalcemia was due to a concurrent decrease in serum PTHrP concentrations, although this seems unlikely. With the availability of the PTHrP immunoassay, it has become possible to control for such unanticipated effects of treatment on PTHrP levels.
Our study hypothesis is that Schussler et al [4] studied the role of calcium in inducing phosphaturia in hypercalcemic breast cancer patients; because it is generally conceded that it is osteolytic bone metastases rather than ectopic PTH secretion that causes hypercalcemia in breast cancer; and concluded that improvement of hypercalcemia after saline infusion is associated with an increase in the TRP independent of ectopic or endogenous PTH secretion. Taking a step further, the present study was designed with the hypothesis of whether the phosphaturia of HHM can be attributed to the hypercalcemia itself rather than a humoral effect of PTHrP secreted by tumor cells.
| Materials and Methods | ▴Top |
A prospective study was done on 20 patients diagnosed with HHM at Icahn School of Medicine at Mount Sinai Services-Queens Hospital Center, Jamaica, New York. All these patients had elevated PTHrP, normal renal function and a low intact-PTH. However, 11 patients were excluded because of early use of drug therapy for hypercalcemia (pamidronate or calcitonin) or radiation therapy or inability to collect urine. Nine patients (four females and five males) who were treated for hypercalcemia with hydration completed the study. Written informed consent was taken from each recruited patient and study design was approved in advance by institutional ethical board review committee.
Patients who had one or more of the following conditions were excluded: elevated serum PTH level, pre-existing renal diseases and medications known to interfere with calcium and bone metabolism including calcium supplement, vitamin D, bisphosphonates, and glucocorticoids. Blood samples were obtained and immediately processed for biochemical analyses before and after specific and supportive treatments for hypercalcemia were initiated. All patients received vigorous intravenous hydration with normal saline at 200 cc/h. After the first 24 h of hydration, small doses of furosemide (20, 40 mg BID) were given for 48 - 72 h in patients number 3 and 7 (Table 1). None of the patients received any other modality of therapy (i.e., pamidronate or calcitonin) for the hypercalcemia during these observations. However, one of our patients (no. 3) was subsequently treated with pamidronate due to poor initial response to hydration and furosemide. Moreover, blood pressure, pulse, and temperature were monitored every 8 h, and questioning regarding any new complaints was conducted several times a day.
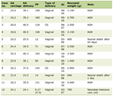 Click to view | Table 1. Variabilities in Biochemical Measurements Before and After Hydration Therapy in Hypercalcemia of Malignancy |
Serum calcium, inorganic phosphate, creatinine, albumin and PTHrP concentrations were measured by standard automated techniques. With the initiation of hydration 24 h urine for creatinine, calcium, and phosphorous were also collected. When the serum calcium had decreased by at least 1.5 mg/dL (usually at 48 - 72 h of hydration), the blood and urine tests were repeated. Serum and urine calcium and phosphate determinations were done by colorimetric assay (Johnson & Johnson); creatinine was measured by two point rate assay; PTHrP and intact-PTH determination was done through immunoradiometric assay (IRMA) by Nicole Institute in San-Juan California and Beckman Coulter respectively [5].
Data were expressed as mean ± standard deviation (SD) or percent as appropriate. Statistical analyses were performed using Statistical Packages for the Social Sciences (SPSS) version 11. A P-value of < 0.05 was considered statistically significant.
| Results | ▴Top |
Table 1 presents the serum value of calcium, phosphate, PTHrP, and TRP before and after hydration therapy. On hydration, a statistically significant (P < 0.001) decline in serum calcium levels had been seen from 13.9 ± 2.1 to 10.8 ± 1.48 mg/dL. Although no significant changes were noticed among phosphate and PTHrP levels, TRP had been statistically (P < 0.05) improved from 69.54±8.16% to 77.85±12.24%. On the exclusion of case no.3, hydration therapy had still shown a statistically significant decrease (13.7 ± 2.1 to 10.5 ± 1.2 mg/dL) in serum calcium (P < 0.001) and increase (70.63±8.00% to 81.21±7.44%) in TRP rate (P < 0.01), respectively.
A statistically significant (P < 0.05) improvement in TRP% has been seen from 69.54 ± 8.16 to 77.85 ± 12.24; however, average value did not reach normal value which might be due to a concurrent improvement in the glomerular filtration rate with the correction of hypercalcemia and consequent increase in TRP did not result in a comparable decrease in phosphate clearance.
Figure 1 represents graphical correlation between serum calcium and TRP levels before and after normal saline therapy, where correction of hypercalcemia improves TRP with statistically no change in the serum concentration of PTHrP (before: 5.20 ± 4.04; after: 5.19 ± 4.03). Thus, it can be stated that the observed increase in TRP was not secondarily significantly related to serum PTHrP levels. Although furosemide was used among two patients, it has been reported by Rodriguez et al that urinary excretion of phosphate is not affected despite a marked diuresis being caused by furosemide [6].
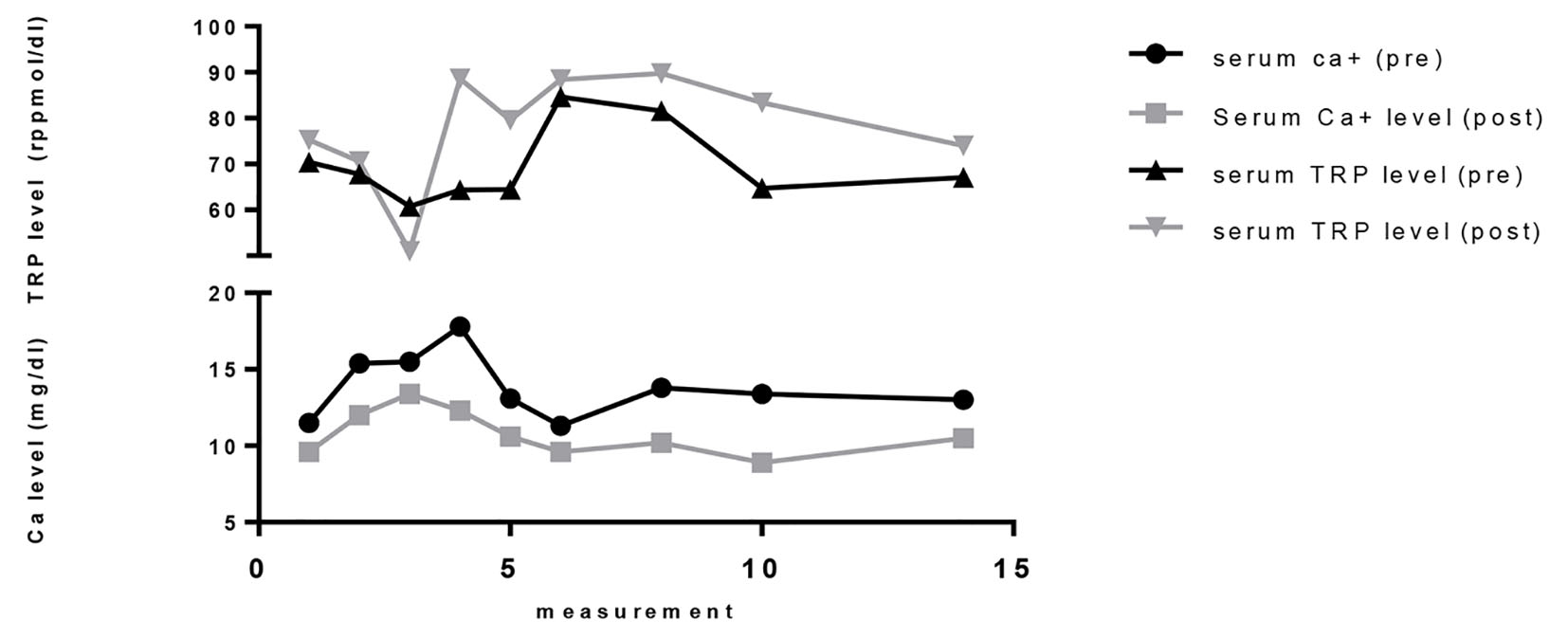 Click for large image | Figure 1. Variabilities in biochemical measurements before and after hydration therapy in hypercalcemia of malignancy. |
| Discussion | ▴Top |
The current study demonstrates that TRP increased toward normal with treatment of hypercalcemia by normal saline therapy in HHM, which explains a phosphaturic effect of hypercalcemia rather than a humoral effect of PTHrP secreted by tumor cells.
Role of calcium in renal phosphate handling
The rate of phosphate transport is dependent on the abundance of three renal sodium phosphate co-transporters functioning, Npt2a, Npt2c, and PiT-2 and the magnitude of the Na+ gradient maintained across the luminal membrane, with the latter depending on the Na+/K+/ATPase or sodium pump on the basolateral membrane. The binding of PTH/PTHrP to parathyroid hormone 1 receptor (PTH1R) produces activation of the Gs/AC/cAMP/PKA and Gq/PLC/IP3/Ca2+/PKC pathways (Fig. 2). The subsequent rise in intracellular calcium suggests that hypercalcemia plays a role in internalization and subsequent degradation of these sodium phosphate co-transporters promotes phosphaturia [7]. Similarly to the current study various authors have proposed several data regarding renal correlation of calcium and phosphate handling. Both Rajagopal et al [8] and Kenny et al [9] have reported mutations in the Npt2a gene that resulted in phenotypes of renal phosphate wasting with resultant hypercalcemia, hypercalciuria, and hypophosphatemia. This suggests that an impaired expression of sodium phosphate co-transporters is associated with dysregulated serum calcium homeostasis as a result of its impact on serum phosphate levels.
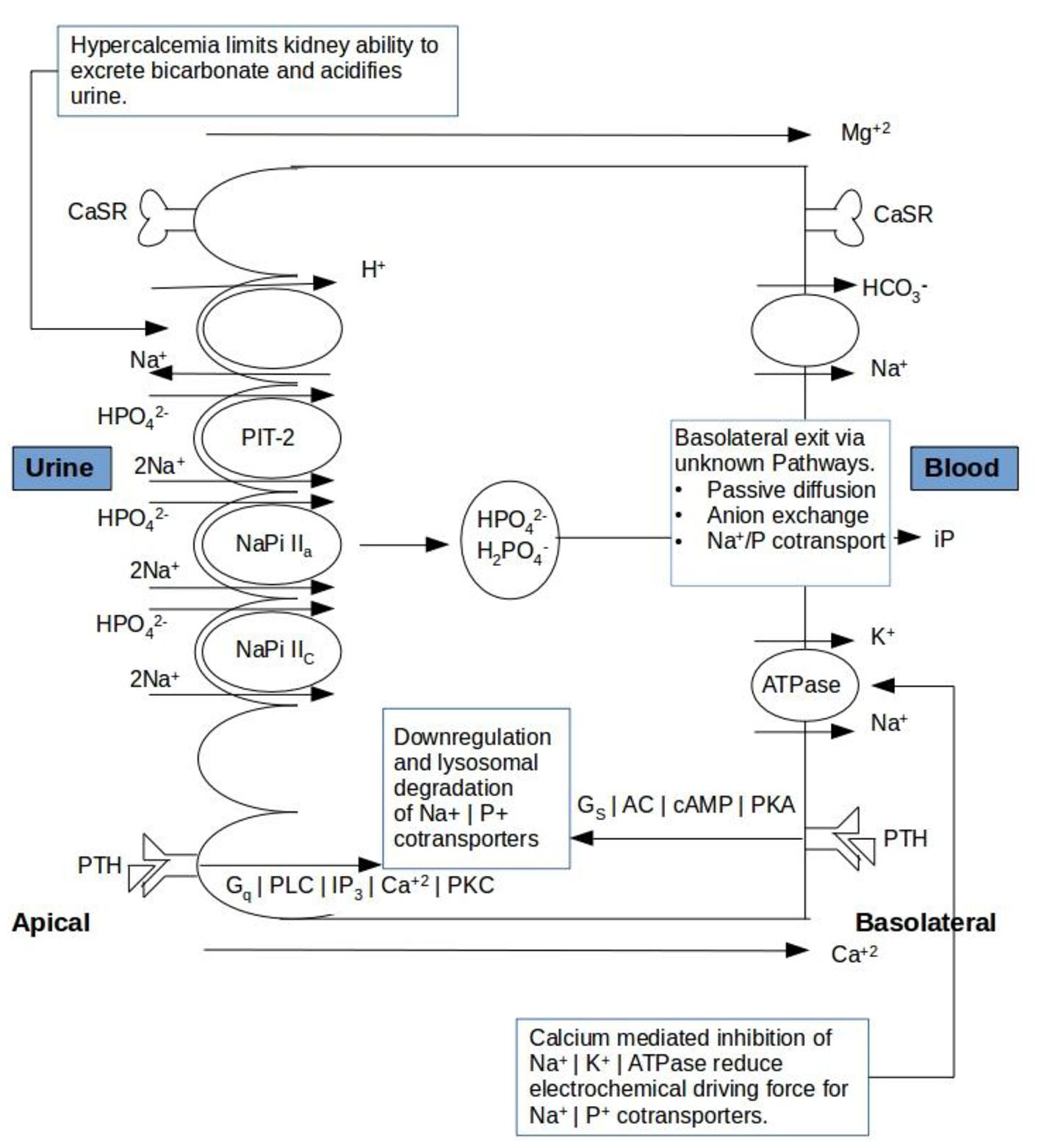 Click for large image | Figure 2. Schematic presentation of calcium mediated regulation of tubular reabsorption of phosphate and bicarbonate ions in renal tubules. CaSR: calcium sensing receptor; PTH: parathyroid hormone. |
Interestingly, Okuda et al reported that the calcium sensing receptor mediates inhibition of Na-ATPase activity in proximal tubular cells [10]. Inhibition of Na/K-ATPase in HHM patients will reduce the electrochemical driving force for Na-PO4 co-transport into the cell which will subsequently result in a decreased TRP (Fig. 1). Although there is no direct evidence of a calcium receptor effect on phosphate transport, it is found that subjects with benign hypocalciuric hypercalcemia due to insensitivity of the calcium receptor show less phosphaturia than patients with the same degree of hypercalcemia due to primary hyperparathyroidism [11]. It will further support our hypothesis that management of hypercalcemia would improve TRP.
Role of calcium in metabolic alkalosis
Patients with advanced cancer manifesting with HHM usually have reduced muscle mass and therefore reduced creatine production and circulating creatinine concentration which potentially interfere with clearance measures, tubular reabsorption calcium index (TRCaI), and fractional excretion of calcium (FECa) calculations. Further, this hypercalcemia with suppressed PTH levels can limit the kidney’s ability to excrete bicarbonate under direct tubular effects on Na/H antiporter and acidifies urine (Fig. 1), which in turn, increases the fraction of ionized calcium along the proximal tubule and promotes a vicious cycle of hypercalcemia [12]. Moreover, hypercalcemia can further explain the increased urinary cyclic AMP since the calcium sensing receptor (like PTH-R) is G protein linked [13].
Effect of PTH and PTHrP on serum calcium levels
PTH stimulates renal calcium reabsorption in the renal tubules [14], and thus contributes pathogenetically in inducing hypercalcemia in HPT; however, controversy surrounds regarding the role of PTHrP in stimulating renal calcium reabsorption in patients with HHM. Some have reported that PTHrP-induced renal calcium reabsorption is unimportant pathophysiologically in HHM [1, 15], whereas others have suggested that the reverse is true [16, 17]. Syed et al [18] had observed that PTHrP-induced renal calcium reabsorption in concert with accelerating osteoclastic bone resorption contributes in a significant way to the hypercalcemia observed in patients with HHM. However, circulation of a larger amino-terminal form(s), a mid-region form(s), and a carboxyl-terminal form(s) which are not fully characterized in structural and functional terms yet may have effects on renal calcium handling that are distinct from those of synthetic PTHrP-(1-36) infusions or injection in HHM [19]. Moreover, it is difficult to assign a quantitative contribution of PTHrP for the role of renal calcium reabsorption compared with that of osteoclastic bone resorption in the pathophysiology of HHM. Thirdly, they infused the study subjects with PTHrP (1-36) to achieve serum PTHrP levels of 8 pmol/kg/h that are much higher than the observed in HHM and among patients except patient number 3 in the present study. Interestingly, Sriussadaporn et al [20] have concluded that serum calcium correlated to serum iPTH more strongly than to plasma PTHrP levels.
The current study does not suggest that PTHrP could not decrease TRP, increase renal calcium reabsorption index, or increase 1a hydroxylation of vitamin D through the dose as higher as 28 pmol/kg/h. These have been described with PTHrP infusion but the plasma concentrations of PTHrP achieved were much higher than observed in HHM including the present study and were usually given for a brief duration [8, 18]. Hence, it has been proposed that the elevated PTHrP frequently associated with HHM in patients without radiologically demonstrable bone metastases are due to PTHrP produced by micrometastases in response to release of transforming growth factor-β (TGF-β) from fertile bone environment [21].
Role of calcium in 1,25-(OH)2D3 levels
It has been claimed that hypercalcemia itself in HHM suppresses 1-α hydroxylation in kidney resulting in suppressed levels of 1,25-(OH)2D3 levels. Bland et al [22] support this statement by showing that cells grown in low calcium (0.5 mmol/L) showed a 4.8-fold induction of 1a-hydroxylase, whereas treatment with medium containing high levels of calcium (2 mmol/L) significantly inhibits 1,25-(OH)2D3 production. Further suggest that direct effects of calcium on proximal tubule cells may be an important feature of the regulation of renal 1,25-(OH)2D3 production. On the other hand, Mosekilde et al [23] have concluded that serum 25-hydroxy vitamin D and renal function are the main determinants for serum 1,25-(OH)2D3 in primary hyperparathyroidism, and showed no significant correlation between serum 1,25-(OH)2D3 and serum calcium, phosphate and PTH, respectively. Hence, we propose that the apparent humoral effects are most readily explained by the hypercalcemia itself.
Our proposal
Thus as demonstrated with the support of other studies, the hypercalcemia can account for decrease in TRP, suppression of PTH levels, low 1,25-(OH)2D3 levels, metabolic alkalosis, and increased cyclic AMP production in HHM irrespective of PTHrP secretion by tumor cells.
Limitations
Though we proposed the effect of PTHrP is from bony micrometastases on serum calcium levels, and defined the inverse correlation between hypercalcemia and TRP, we could not estimate the magnitude of influence of various other cytokines such as IL-6, tumor necrosis factor, IL-1 or TGF-a produced in patients with malignancies. Presumably, these cytokines act at different sites and play a significant role in paraneoplastic leukocytosis, cachexia, and further potentiate the effects of PTHrP on the osteoclast lineages. Moreover, the current study could not distinguish the role of various PTH independent phosphatonins, including fibroblast growth factor, secreted frizzled-related protein-4, dentin matrix protein 1 and matrix extracellular phosphoglycoprotein, which had shown an influence on TRP [24].
Conclusion
We suggest as per the results of the present study that the humoral effects of PTHrP in HHM can be explained by hypercalcemia itself, state that improvement of hypercalcemia by hydration reverses phosphaturia without reducing PTHrP levels, and further imply that most cases of HHM associated with elevated PTHrP could be the effect from uncoupled bone formation-resorption rate through a local autocrine or paracrine action produced by non-radiographically demonstrated bony micrometastases rather than a humoral hypercalcemia.
Acknowledgments
We acknowledge Dr. Paul Kim for his contribution in a graphical presentation of the study results, and Dr. Jaspinder Kaur for her efforts in doing all the literature research, and designing and writing the manuscript for this study. We are grateful to our late mentor Dr. George Schussler.
Conflicts of Interest
None.
Financial Support
None.
| References | ▴Top |
- Stewart AF, Horst R, Deftos LJ, Cadman EC, Lang R, Broadus AE. Biochemical evaluation of patients with cancer-associated hypercalcemia: evidence for humoral and nonhumoral groups. N Engl J Med. 1980;303(24):1377-1383.
doi pubmed - Mundy GR. Pathogenesis of hypercalcaemia of malignancy. Clin Endocrinol (Oxf). 1985;23(6):705-714.
doi - Lavender AR, Pullman TN. Changes in inorganic phosphate excretion induced by renal arterial infusion of calcium. Am J Physiol. 1963;205(5):1025-1032.
pubmed - Schussler GC, Verso M, Nemoto T. Phosphaturia in hypercalcemic breast cancer patients. J Clin Endocrinol Metab. 1972;35(4):497-504.
doi pubmed - Prasad UV, Mohan RK, Samuel G, Harinarayan CV, Sivaprasad N, Venkatesh M. Standardisation of a two-site PTH immunoradiometric assay using various solid phase formats. Indian J Med Res. 2012;136(6):963-970.
pubmed - Rodriguez HJ, Walls J, Yates J, Klahr S. Effects of acetazolamide on the urinary excretion of cyclic AMP and on the activity of renal adenyl cyclase. J Clin Invest. 1974;53(1):122-130.
doi pubmed - Murray RD, Lederer ED, Khundmiri SJ. Role of PTH in the Renal Handling of Phosphate. Medical Science. 2015;2(3):162-181.
doi - Rajagopal A, Braslavsky D, Lu JT, Kleppe S, Clement F, Cassinelli H, Liu DS, et al. Exome sequencing identifies a novel homozygous mutation in the phosphate transporter SLC34A1 in hypophosphatemia and nephrocalcinosis. J Clin Endocrinol Metab. 2014;99(11):E2451-2456.
doi pubmed - Kenny J, Lees MM, Drury S, Barnicoat A, Van't Hoff W, Palmer R, Morrogh D, et al. Sotos syndrome, infantile hypercalcemia, and nephrocalcinosis: a contiguous gene syndrome. Pediatr Nephrol. 2011;26(8):1331-1334.
doi pubmed - Okuda M, Cheng S, Hall AE, Herbert SC. Calcium - Sensing Receptor Mediates the Inhibition of Na+, K+ Atphase Activity in the Proximal tubular cells. Am Neph Society. 2000 (A3000) (Abstract).
- Attie MF, Gill JR, Jr., Stock JL, Spiegel AM, Downs RW, Jr., Levine MA, Marx SJ. Urinary calcium excretion in familial hypocalciuric hypercalcemia. Persistence of relative hypocalciuria after induction of hypoparathyroidism. J Clin Invest. 1983;72(2):667-676.
doi pubmed - Wagner CA. The calcium-sensing receptor directly regulates proximal tubular functions. Kidney Int. 2013;84(2):228-230.
doi pubmed - Hebert SC, Brown EM, Harris HW. Role of the Ca(2+)-sensing receptor in divalent mineral ion homeostasis. J Exp Biol. 1997;200(Pt 2):295-302.
pubmed - Bushinsky DA. Calcium, magnesium and phosphorus: renal handling and urinary excretion. In: Favus M, ed. Primer on the metabolic bone diseases and disorders of mineral metabolism. Philadelphia: Lippincott, Williams & Wilkins; 1999; p. 67-74.
- Godsall JW, Burtis WJ, Insogna KL, Broadus AE, Stewart AF. Nephrogenous cyclic AMP, adenylate cyclase-stimulating activity, and the humoral hypercalcemia of malignancy. Recent Prog Horm Res. 1986;42:705-750.
doi - Buchs B, Rizzoli R, Bonjour JP. Evaluation of bone resorption and renal tubular reabsorption of calcium and phosphate in malignant and nonmalignant hypercalcemia. Bone. 1991;12(1):47-56.
doi - Ralston SH, Boyce BF, Cowan RA, Gardner MD, Fraser WD, Boyle IT. Contrasting mechanisms of hypercalcemia in patients with early and advanced humoral hypercalcemia of malignancy. J Bone Miner Res. 1989;4(1):103-111.
doi pubmed - Syed MA, Horwitz MJ, Tedesco MB, Garcia-Ocana A, Wisniewski SR, Stewart AF. Parathyroid hormone-related protein-(1 - 36) stimulates renal tubular calcium reabsorption in normal human volunteers: implications for the pathogenesis of humoral hypercalcemia of malignancy. J Clin Endocrinol Metab. 2001;86(4):1525-1531.
pubmed - Burtis WJ, Dann P, Gaich GA, Soifer NE. A high abundance midregion species of parathyroid hormone-related protein: immunological and chromatographic characterization in plasma. J Clin Endocrinol Metab. 1994;78(2):317-322.
pubmed - Sriussadaporn S, Phoojaroenchanachai M, Ploybutr S, Plengvidhya N, Peerapatdit T, Nitiyanant W, Vannasaeng S, et al. Hypercalcemia of malignancy: a study of clinical features and relationships among circulating levels of calcium, parathyroid hormone and parathyroid hormone-related peptide. J Med Assoc Thai. 2007;90(4):663-671.
pubmed - Guise TA. Molecular mechanisms of osteolytic bone metastases. Cancer. 2000;88(12 Suppl):2892-2898.
doi - Bland R, Walker EA, Hughes SV, Stewart PM, Hewison M. Constitutive expression of 25-hydroxyvitamin D3-1alpha-hydroxylase in a transformed human proximal tubule cell line: evidence for direct regulation of vitamin D metabolism by calcium. Endocrinology. 1999;140(5):2027-2034.
- Mosekilde L, Charles P, Lindegreen P. Determinants for serum 1,25-dihydroxycholecalciferol in primary hyperparathyroidism. Bone Miner. 1989;5(3):279-290.
doi - Bacic D, Lehir M, Biber J, Kaissling B, Murer H, Wagner CA. The renal Na+/phosphate cotransporter NaPi-IIa is internalized via the receptor-mediated endocytic route in response to parathyroid hormone. Kidney Int. 2006;69(3):495-503.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.