

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website http://www.jofem.org |
Case Report
Volume 5, Number 4, August 2015, pages 261-266
Cushing Syndrome Due to Primary Pigmented Nodular Adrenal Disease in Three Related Adolescent Girls With Carney Complex
Beatriz Fragaa, Catarina Francoa, Sara Vaza, Fernanda Gomesa, Margarida Sa-Fernandesb, Jorge Pinheirob, Jose Estevao-Costac, John Aidan Carneyd, Joao Anselmoe, f
aDepartment of Pediatrics, Hospital Divino Espirito Santo, Ponta Delgada, Acores, Portugal
bDepartment of Pathology, Hospital S. Joao, Porto, Portugal
cDepartment of Pediatric Surgery, Faculty of Medicine, Hospital S. Joao, Porto, Portugal
dDepartment of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN 55902, USA
eDepartment of Endocrinology, Hospital Divino Espirito Santo, Ponta Delgada, Acores, Portugal
fCorresponding Author: Joao Anselmo, Department of Endocrinology, Hospital Divino Espirito Santo, 9500-317 Ponta Delgada, Acores, Portugal
Manuscript accepted for publication August 14, 2015
Short title: PPNAD and Puberty
doi: http://dx.doi.org/10.14740/jem299w
| Abstract | ▴Top |
Primary pigmented nodular adrenal disease (PNNAD) is a rare cause of Cushing syndrome particularly during childhood and adolescence. Females are preferentially affected. The present work is aimed at assessing if there is a relationship between puberty and the development of Cushing syndrome due to PPNAD. We have followed an extended family with Carney complex (CNC) due to a dominantly inherited mutation of the gene encoding the regulatory subunit 1α (R1A) of the protein kinase A (PRKAR1A). The large majority of family members presenting with Cushing syndrome due to PPNAD were women (six out of seven). One of them died of an adrenocortical carcinoma at age 27 years. Genetic screening of entire family allowed follow-up of those family members harboring the mutation but without clinical manifestations of hypercortisolism. During the last 2 years, three related adolescent girls developed subclinical hypercortisolism starting shortly after menarche. None of their male relatives carrying the same mutation showed abnormal adrenal function. The first abnormality noticed in the patients was loss of the normal circadian variation of cortisol production. A low dose dexamethasone test (Liddle’s test) elicited a paradoxical increase in urinary excretion of free cortisol greater than 50% of basal values. Several months later, they complained of insidious manifestations of hypercortisolism including weight gain, hypertension and oligomenorrhea. They underwent laparoscopic bilateral adrenalectomy. Pathologically, the excised glands showed PPNAD. We conclude that female gender is a predisposing factor for expressing PPNAD. A paradoxical rise of urinary free cortisol (UFC) in response to dexamethasone is an early and specific laboratory marker of the disorder, allowing timely bilateral adrenalectomy which may not only prevent severe complications of hypercortisolism, but may also preclude the development of adrenocortical carcinoma.
Keywords: Carney complex; Primary pigmented nodular adrenal disease; Cushing syndrome; Menarche; Glucocorticoids
| Introduction | ▴Top |
Cushing syndrome in childhood and adolescence may present difficult diagnostic and therapeutic challenges. Hypercortisolism can be classified as adrenocorticotropin (ACTH)-dependent (Cushing disease) and ACTH-independent (Cushing syndrome). The latter type includes some hereditary disorders, one of which is primary pigmented nodular adrenocortical disease (PPNAD) which occurs as a sporadic disorder or as part of the Carney complex (CNC) [1]. CNC is a multiple endocrine neoplasia syndrome with dominant inheritance and involvement of different organs and tissues particularly endocrine glands (pituitary and gonads in addition to adrenal) and the skin (lentigines and myxomas) [2]. Defects of the regulatory subunit 1α (R1A) of the protein kinase A (PRKAR1A) gene have been identified in 62% of the affected patients [3, 4]. PPNAD is the most common endocrine manifestation of CNC occurring in about 60% of affected patients [5]. However, in virtually all necropsy studies of patients with CNC, PPNAD features were identified in the adrenal glands, suggesting that in some cases the disease might have an indolent course [6]. Since Cushing syndrome develops slowly over several years and its clinical manifestations may be subtle [7, 8], establishing the diagnosis of PPNAD is sometimes difficult. Furthermore, cyclic or episodic forms of Cushing syndrome may occur with PPNAD [9, 10]. It is therefore not always easy to trace the onset of hypercortisolism to any particular event.
We have followed an extended family with Carney complex. Six of seven affected members with Cushing syndrome due to PPNAD were women [11]. Genetic screening of the entire family allowed us to follow those members of the family harboring the mutation for manifestations of hypercortisolism. In view of the fact that the first alterations in adrenal function in the affected patients started shortly after menarche and that none of the male relatives carrying the same mutation presented abnormalities of the adrenal function, it seemed possible that female sex steroid hormones played an important role in the development of PPNAD.
| Case Report | ▴Top |
During the last 10 years, we have followed an extended Azorean family with CNC (Fig. 1). Multiple members, mostly women (six of seven), developed Cushing syndrome due to PPNAD which was diagnosed during their third decade. In view of the high prevalence of PPNAD in this family, sequencing of the coding region of the PRKAR1A gene was carried out in all family members. A heterozygous mutation in the PRKAR1A gene, consisting of a single nucleotide change (c439A>G) which leads to substitution of glycine with arginine at the position 147 (S147G) of the R1A of the cAMP-dependent protein kinase A (PKA) was identified in 15 family members. This mutation segregated with the disease and was transmitted in an autosomal dominant manner. All family members with the mutation were followed biannually in our outpatient clinic. At each visit, clinical manifestations of CNC were carefully checked and a routine panel of blood tests was obtained. In addition, the following hormonal determinations were obtained six-monthly: prolactin, estradiol, luteinizing hormone (LH), follicle stimulating hormone (FSH), insulin-like growth factor 1 (IGF-1); free thyroxine (FT4) and thyrotropin (TSH); ACTH and cortisol (morning and late-afternoon); 24-h urinary cortisol and creatinine excretions; serum dehydroepiandrosterone sulfate (DHEA-S); androstenedione and 17-α-hydroxyprogesterone were determined annually. Magnetic resonance imaging (MRI) of the pituitary and CT of the thyroid, adrenal glands and ovaries were performed when indicated. An echocardiogram was carried out at the beginning of the follow-up. When laboratory tests of adrenal function were found to be abnormal (loss of circadian variation and/or increased urinary free cortisol (UFC) excretion), the patient was admitted to the hospital for a low dose (2 mg) dexamethasone test (Liddle’s test). Blood samples were drawn in the morning and at midnight just before and after administration of oral dexamethasone, 0.5 mg each 6 h for 48 h. The 24-h urine specimens were collected during three consecutive days.
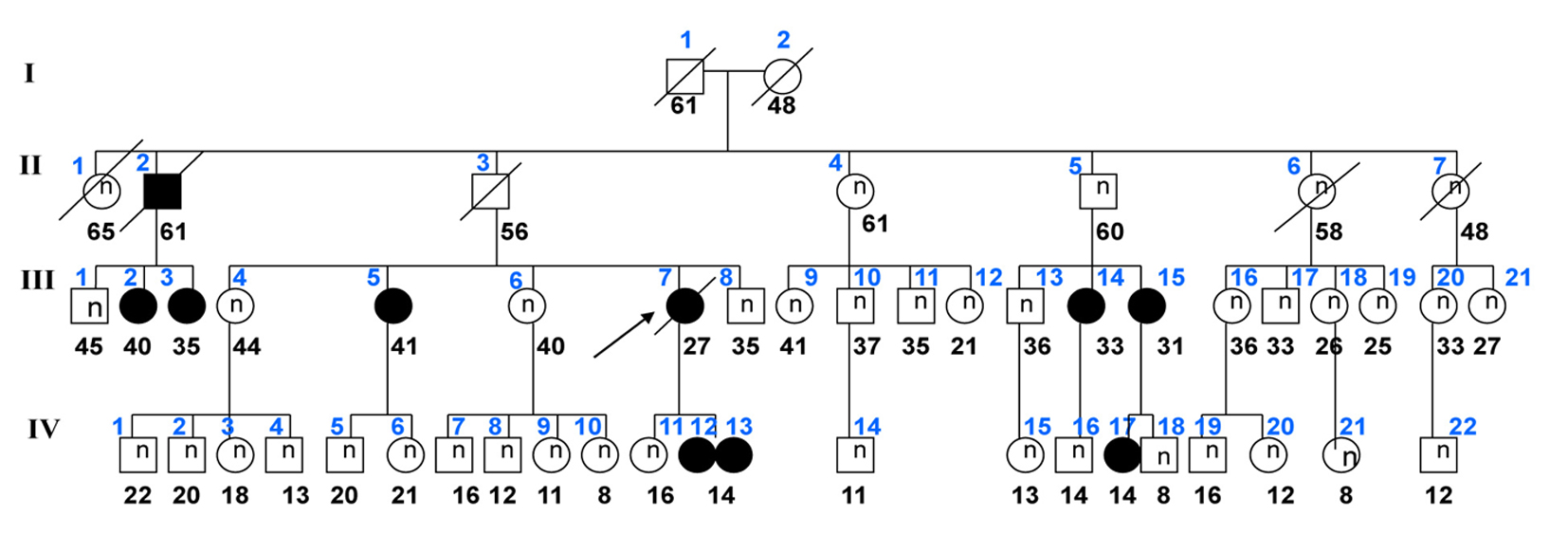 Click for large image | Figure 1. Genealogic tree of the family. Arrow indicates the index case. Circles identify female members; squares indicate males; black symbols show patient with PPNAD. Age of the subjects is shown below the symbols. Family members are numbered by generation above symbol. Examined members who are normal (suppressed cortisol in response to 1 mg of dexamethasone) are identified with (n) inside the symbol. |
Three adolescent girls of the family (monozygotic twins and their second-degree cousin) with PRKAR1A mutation developed laboratory and clinical manifestations of Cushing syndrome starting after onset of menarche (Table 1). The monozygotic twin sisters (cases 1 and 2) but not their cousin (case 3) had spotty skin pigmentation involving the vermilion border of the lips, eyelids, conjunctiva and the genital mucosa (Fig. 2). The pigmentation dated from birth and became darker around the puberty. One (case 2) also had blue nevi. Other manifestations of CNC, in particular cardiac myxomas, ovary tumors, thyroid nodules and cutaneous myxomas were not found. The patients’ development was normal. Menarche occurred around the age of 12 years (Table 1). Menstrual cycles were regular for the first year; then oligo/amenorrhea, hypertension and rapid weight gain (> 5 kg/year) were noticed. Previously, ACTH and serum and urinary cortisol levels were within the normal range (Table 2). Laboratory alterations in adrenocortical function were detected around the age of 12 years and included absence of circadian variation of serum cortisol levels (morning and afternoon cortisol values were similar). At this time, late-night cortisol was above 5 µg/dL. ACTH was low in cases 1 and 2, and normal in case 3. During Liddle’s test, dexamethasone administration did not suppress serum cortisol; on the contrary, it elicited a paradoxical increase in urinary cortisol excretion greater than 50% of the basal values (Table 2). CT scans of the adrenal glands were normal.
 Click to view | Table 1. Clinical Features Before Bilateral Adrenalectomy |
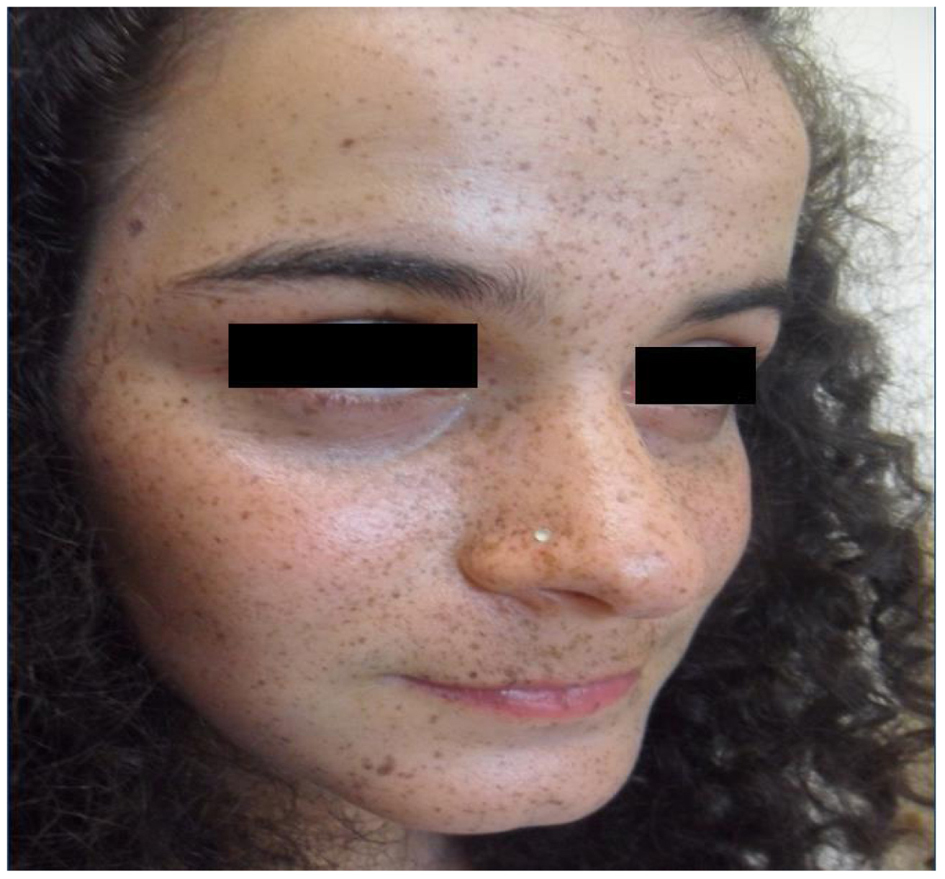 Click for large image | Figure 2. Centrofacial lentigines including the vermilion borders of the lips. |
Click to view | Table 2. Evolution of Adrenal Function in Affected Patients |
Bilateral laparoscopic adrenalectomy was performed in all three patients. The adrenal glands were normal-sized and exhibited dark nodules. Microscopically, the nodules were composed of large cortical cells with eosinophilic cytoplasm; the nuclei were large and hyperchromatic with prominent nucleoli; the cytoplasm was rich in lipofuscin. The non-nodular cortex was atrophic (Fig. 3).
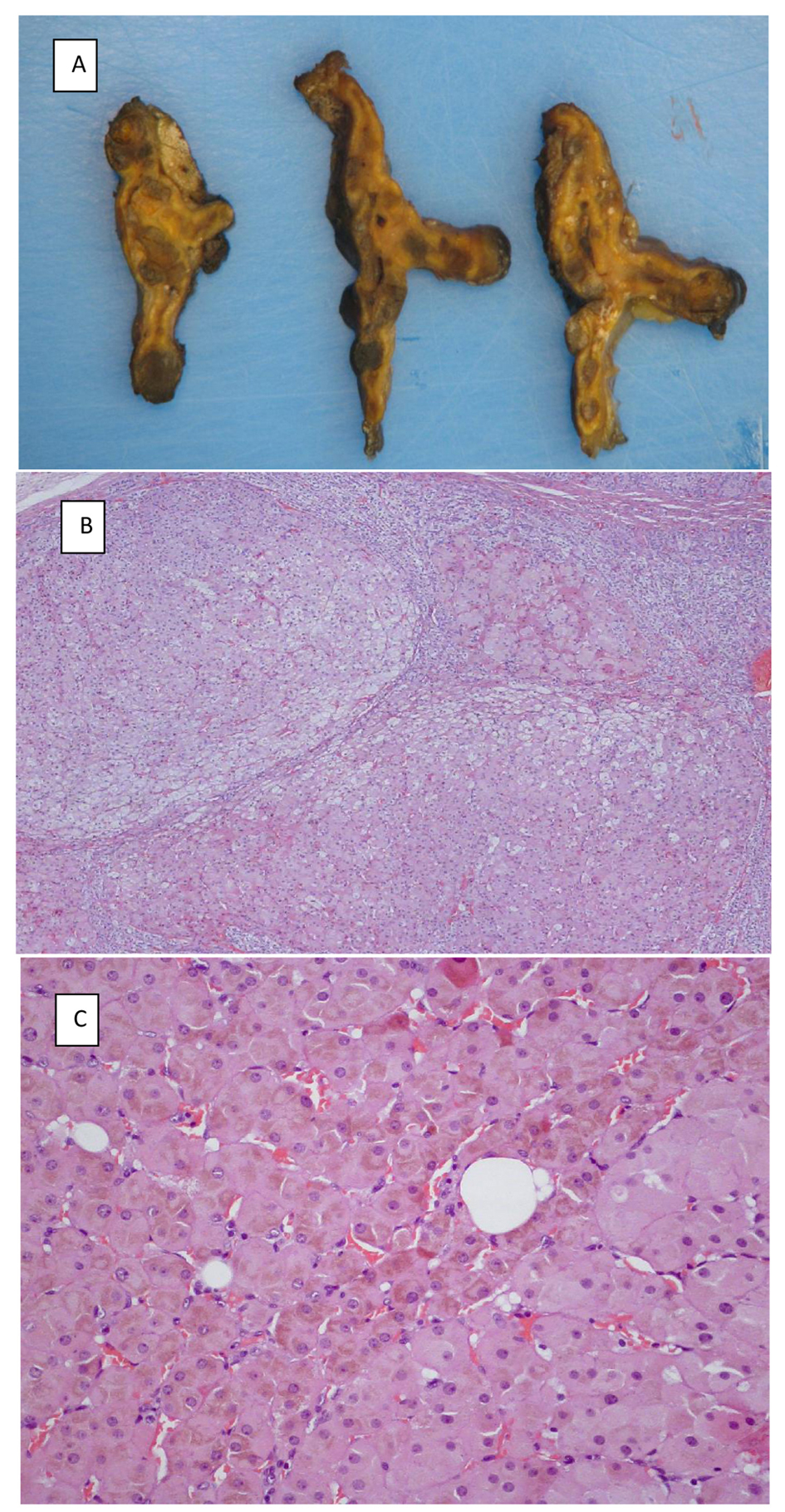 Click for large image | Figure 3. (A) Macroscopic appearance of transverse slices of the left adrenal gland removed from patient IV-17. (B) Adrenal nodules surrounded by atrophic cortex. (C) Large cortical cells with eosinophilic cytoplasm rich in lipofuscin. |
Postoperatively, oral hydrocortisone was administered in two divided doses adjusted to mimic the natural circadian variation of cortisol secretion. The daily dose was on average 9.6 mg/m2. Fludrocortisone was given in the morning based on body weight (average of 50 µg daily). Within 3 months, menses resumed, blood pressure returned to normal levels and patients lost weight.
| Discussion | ▴Top |
In the present work we found, for the first time, a close relationship between the menarche and onset of hypercortisolism in three adolescent girls belonging to a family with CNC and PRKAR1A mutation. This was possible because genetic screening of the entire family allowed us to follow carriers of the mutation before laboratory and clinical manifestations of hypercortisolism became evident. Most of the family members (nine out of 10) with clinical Cushing due to PPNAD were female (a total of 15 family members had the mutation).
In the three cases herein described, clinical and laboratory manifestations of hypercortisolism started shortly after menarche raising the possibility that female sex steroid hormones might be linked to the expression of the disorder, at least in this family. Indeed, PPNAD nodules are known to overexpress progesterone and estradiol receptors [12, 13]. Moreover, in older patients of this family with Cushing syndrome, clinical and laboratory manifestations of hypercortisolism were greatly intensified during pregnancy, reinforcing the influence of female sex steroid hormones in triggering autonomous production of cortisol. Overactivity of adrenal steroidogenesis in the early stage of PPNAD is restricted to cortisol. In fact, serum DHEA-S, an androgenic precursor exclusively produced by the adrenal gland was normal in our three patients (Table 2), albeit with a progressive increase with age related to adrenarche. Levels of DHEA-S were increased only in the family member with adrenocortical carcinoma.
The paradoxical rise in UFC excretion induced by dexamethasone proved to be an early laboratory marker of PPNAD. The finding has been reported as being diagnostic of the disorder [14]. In a study that compared 21 patients with PPNAD with others with adrenal adenoma and macronodular adrenal hyperplasia, an increase in UFC greater than 50%, identified 70% of the cases of PPNAD. An increase in UFC of more than 100% over basal values, distinguished virtually all patients with PPNAD from patients with other primary adrenocortical disorders [15]. In our patients, UFC rose by more than 50% (range 50-159%) in response to a low dose dexamethasone test (Liddle’s test). Since these patients had a PRKAR1A gene mutation, a paradoxical increase in UFC made the diagnosis of PPNAD almost certain. Dexamethasone administration did suppress cortisol secretion in their male relatives harboring the same mutation (Fig. 1; members II-5, III-13, IV-5 and IV-18). This finding supports the notion of an important role for female sex steroid hormones in the expression of PPNAD. The molecular mechanisms involved in dexamethasone-induced cortisol secretion have not been fully elucidated. The paradoxical response to dexamethasone also occurred in vitro [16], pointing to an intrinsic defect in the adrenal cortisol-producing cells. Similar to the receptors of female steroid hormones, glucocorticoid receptors (GRs) were also found to be overexpressed in PPNAD nodules whereas they were barely detectable in the internodular cortex. The stimulating effect of dexamethasone seems to be mediated by PKA; however, other pathways may also be involved [17]. In addition to Cushing syndrome, the mother of our twin sisters (Fig. 1; patient II-7) died of an adrenocortical carcinoma arising in a gland with PPNAD features [11]. It is still not clear if PPNAD has malignant potential; one other such case has been described [18]. The possibility of that PPNAD may favor adrenocortical carcinogenesis cannot be disregarded [19, 20].
In conclusion, we have shown that hypercortisolism due to PPNAD in three closely related adolescents with PRKAR1A mutations started shortly after menarche, suggesting an influence of female sex steroids on the nodular adrenal cortical development. A paradoxical increase in urinary cortisol excretion is an early marker of the autonomous production of cortisol in PPNAD. Timely bilateral adrenalectomy prevents the severe complications of Cushing syndrome and may also preclude the occurrence of an adrenocortical carcinoma arising in the context of PPNAD.
Acknowledgement
We thank all members of the family for their willingness to participate in this study. We are also grateful to Dr. Constantine Stratakis (NIH, Bethesda) for his contribution to the genetic screening of the family.
Funding Source
No funding was secured for this study.
Financial Disclosure
The authors have no financial relationship relevant to this article to disclose.
Conflict of Interest
There is no conflict of interest to disclose
Abbreviations
PPNAD: primary pigmented nodular adrenocortical disease; CNC: Carney complex; PRKAR1A: protein kinase A (PKA) regulatory subunit 1α (R1A); ACTH: adrenocorticotropin; UFC: urinary free cortisol
| References | ▴Top |
- Stratakis CA, Kirschner LS, Carney JA. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab. 2001;86(9):4041-4046.
doi pubmed - Stratakis CA. Cushing syndrome caused by adrenocortical tumors and hyperplasias (corticotropin- independent Cushing syndrome). Endocr Dev. 2008;13:117-132.
doi pubmed - Kirschner LS, Sandrini F, Monbo J, Lin JP, Carney JA, Stratakis CA. Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the carney complex. Hum Mol Genet. 2000;9(20):3037-3046.
doi pubmed - Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, Cho-Chung YS, et al. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26(1):89-92.
doi pubmed - Bertherat J, Horvath A, Groussin L, Grabar S, Boikos S, Cazabat L, Libe R, et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5'-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab. 2009;94(6):2085-2091.
doi pubmed - Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VL. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine (Baltimore). 1985;64(4):270-283.
doi - Groussin L, Horvath A, Jullian E, Boikos S, Rene-Corail F, Lefebvre H, Cephise-Velayoudom FL, et al. A PRKAR1A mutation associated with primary pigmented nodular adrenocortical disease in 12 kindreds. J Clin Endocrinol Metab. 2006;91(5):1943-1949.
doi pubmed - Stratakis CA. Cushing syndrome in pediatrics. Endocrinol Metab Clin North Am. 2012;41(4):793-803.
doi pubmed - Gunther DF, Bourdeau I, Matyakhina L, Cassarino D, Kleiner DE, Griffin K, Courkoutsakis N, et al. Cyclical Cushing syndrome presenting in infancy: an early form of primary pigmented nodular adrenocortical disease, or a new entity? J Clin Endocrinol Metab. 2004;89(7):3173-3182.
doi pubmed - Powell AC, Stratakis CA, Patronas NJ, Steinberg SM, Batista D, Alexander HR, Pingpank JF, et al. Operative management of Cushing syndrome secondary to micronodular adrenal hyperplasia. Surgery. 2008;143(6):750-758.
doi pubmed - Anselmo J, Medeiros S, Carneiro V, Greene E, Levy I, Nesterova M, Lyssikatos C, et al. A large family with Carney complex caused by the S147G PRKAR1A mutation shows a unique spectrum of disease including adrenocortical cancer. J Clin Endocrinol Metab. 2012;97(2):351-359.
doi pubmed - Caticha O, Odell WD, Wilson DE, Dowdell LA, Noth RH, Swislocki AL, Lamothe JJ, et al. Estradiol stimulates cortisol production by adrenal cells in estrogen-dependent primary adrenocortical nodular dysplasia. J Clin Endocrinol Metab. 1993;77(2):494-497.
pubmed - de Cremoux P, Rosenberg D, Goussard J, Bremont-Weil C, Tissier F, Tran-Perennou C, Groussin L, et al. Expression of progesterone and estradiol receptors in normal adrenal cortex, adrenocortical tumors, and primary pigmented nodular adrenocortical disease. Endocr Relat Cancer. 2008;15(2):465-474.
doi pubmed - Batista DL, Riar J, Keil M, Stratakis CA. Diagnostic tests for children who are referred for the investigation of Cushing syndrome. Pediatrics. 2007;120(3):e575-586.
doi pubmed - Stratakis CA, Sarlis N, Kirschner LS, Carney JA, Doppman JL, Nieman LK, Chrousos GP, et al. Paradoxical response to dexamethasone in the diagnosis of primary pigmented nodular adrenocortical disease. Ann Intern Med. 1999;131(8):585-591.
doi pubmed - Bourdeau I, Lacroix A, Schurch W, Caron P, Antakly T, Stratakis CA. Primary pigmented nodular adrenocortical disease: paradoxical responses of cortisol secretion to dexamethasone occur in vitro and are associated with increased expression of the glucocorticoid receptor. J Clin Endocrinol Metab. 2003;88(8):3931-3937.
doi pubmed - Louiset E, Stratakis CA, Perraudin V, Griffin KJ, Libe R, Cabrol S, Feve B, et al. The paradoxical increase in cortisol secretion induced by dexamethasone in primary pigmented nodular adrenocortical disease involves a glucocorticoid receptor-mediated effect of dexamethasone on protein kinase A catalytic subunits. J Clin Endocrinol Metab. 2009;94(7):2406-2413.
doi pubmed - Morin E, Mete O, Wasserman JD, Joshua AM, Asa SL, Ezzat S. Carney complex with adrenal cortical carcinoma. J Clin Endocrinol Metab. 2012;97(2):E202-206.
doi pubmed - Bossis I, Voutetakis A, Bei T, Sandrini F, Griffin KJ, Stratakis CA. Protein kinase A and its role in human neoplasia: the Carney complex paradigm. Endocr Relat Cancer. 2004;11(2):265-280.
doi pubmed - Groussin L, Kirschner LS, Vincent-Dejean C, Perlemoine K, Jullian E, Delemer B, Zacharieva S, et al. Molecular analysis of the cyclic AMP-dependent protein kinase A (PKA) regulatory subunit 1A (PRKAR1A) gene in patients with Carney complex and primary pigmented nodular adrenocortical disease (PPNAD) reveals novel mutations and clues for pathophysiology: augmented PKA signaling is associated with adrenal tumorigenesis in PPNAD. Am J Hum Genet. 2002;71(6):1433-1442.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.