

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website http://www.jofem.org |
Original Article
Volume 5, Number 4, August 2015, pages 229-237
Autonomic Diabetic Neuropathy Impairs Glucose and Dipeptidyl Peptidase 4 Inhibitor-Regulated Glucagon Concentration in Type 1 Diabetic Patients
Emilie Lobineta, b, c, Francois Reichardta, b, Celine Garreta, b, Laurent Cazalsc, Aurelie Wageta, b, Sylvie Dejajerd, Florence Labrousseb, c, Jean Michel Senarda, b, e, Jens Juul Holstf, Helene Hanaireb, c, g, Remy Burcelina, b, g
aInstitut National de la Sante et de la Recherche Medicale (INSERM), Toulouse, France
bUniversite de Toulouse Paul Sabatier (UPS), Unite Mixte de Recherche (UMR) 1048, Institut des Maladies Metaboliques et Cardiovasculaires (I2MC), F-31432 Toulouse Cedex 4, France
cDepartment of Diabetology Hopital Rangueil, CHU de Toulouse, France
dNovartis Pharma SAS, Rueil Malmaisson, France
eINSERM U1048, Eq08 et Service de Pharmacologie Clinique, CHU de Toulouse, France
fNNF Center for Basic Metabolic Research and Department of Biomedical Sciences, University of Copenhagen, Denmark
gCorresponding Author: Helene Hanaire, Universite de Toulouse Paul Sabatier (UPS), Unite Mixte de Recherche (UMR) 1048, Institut des Maladies Metaboliques et Cardiovasculaires (I2MC), F-31432 Toulouse Cedex 4, France; Remy Burcelin, INSERM 1048 and Helene Hanaire, Hopital Rangueil, 31400 Toulouse, France
Manuscript accepted for publication June 26, 2015
Short title: Neuropathy Hampers Action of Gliptin
doi: http://dx.doi.org/10.14740/jem289w
| Abstract | ▴Top |
Background: Dipeptidyl peptidase 4 inhibitors (DPP4i) could exert their glucagonostatic action through a functional autonomic nervous system independently from insulin secretion. We explored this hypothesis.
Methods: We studied C-peptide negative type 1 diabetic patients (T1D) with (AN+) or without (AN-) autonomic neuropathy. Plasma glucagon, active and total glucagon-like peptide-1 (GLP-1), GIP, pancreatic polypeptide (PP), and glucose concentrations were quantified over 180 minutes following a meal test. Furthermore, to increase the plasma concentration of GLP-1 between groups and study its impact on glucagon secretion, 50 mg of vildagliptin and a second meal test were administered to the same patients.
Results: The plasma concentration of glucagon was higher in AN+ patients than in AN- patients, which was associated with lower PP and active GLP-1 plasma concentrations. This first set of data suggest that AN, presumably involving parasympathetic activity, results in loss of glucose-regulated glucagon secretion. After DPP4i treatment, AN+ patients lost the ability to suppress plasma glucagon, and the low plasma PP responses were not restored.
Conclusions: We here show that a functional autonomic nervous system is required for the proper control of glucagon secretion. The mechanism is insulin-independent. The glucagonostatic action of DPP4i also requires this mechanism.
Keywords: Neuropathy; Gut brain axis; Incretins; GLP-1; Type 1 diabetes
| Introduction | ▴Top |
Elevated or inappropriately high concentrations of glucagon in plasma, relative to plasma glucose and insulin concentrations, characterize type 1 (T1DM) and type 2 diabetic (T2DM) patients. Glucagon-like peptide-1 (GLP-1) exerts a glucagonostatic effect in a glucose-dependent manner meaning that there is no inhibition of glucagon responses to hypoglycemia, which means that GLP-1 does not impair the role of glucagon in the defense against hypoglycemia when it is used therapeutically. This might suggest that the inhibitory mechanism could involve insulin secretion, but it is well established that GLP-1 as well as dipeptidyl peptidase 4 inhibitors (DPP4i) lowers glucagon secretion also in T1D patients with no residual beta cell function, indicating that the effect of GLP-1 on glucagon secretion is mediated, at least in this situation, by a mechanism independent from insulin secretion [1-5]. Such glucose dependent and insulin independent effects could involve neural mechanisms, since the plasma glucagon concentration is under the control of glucose-sensitive neural units mostly located within the brain [6, 7]. These units appear to be able to detect the glycemic variations and signal to the pancreatic alpha cells to enhance or reduce secretion. Some of the molecular mechanisms have been elucidated and appear to involve the glucose transporter GLUT2 [7], the inward rectifying potassium channel Kir 6.2 [8, 9] and perhaps the brain GLP-1 receptor [10]. Regarding GLP-1, an endocrine regulation directly on the pancreatic alpha cells for the control of glucagon secretion would seem incompatible with the rapid degradation of GLP-1 by the DPP4 which means that very little GLP-1 reaches the pancreas in the intact form [11]. Furthermore, whether or not the GLP-1 receptor on pancreatic alpha cell is responsible for the glucagonostatic effect of GLP-1 remains highly controversial. For similar reasons, a mechanism involving stimulation by GLP-1 of somatostatin secreted from neighboring delta cells, which can be demonstrated in the perfused rat pancreas [12], may be relevant only for GLP-1 agonist administered in high doses. Together, this has called for other, non-endocrine, hypotheses. Our hypothesis is that enteric GLP-1 might activate a gut-to brain-to-pancreatic alpha cell neuronal axis to control glucagon secretion during a meal. This hypothesis might help to explain the glucagonostatic effect of the DPP4i, since they are known to augment the concentration of active GLP-1 in the blood of the hepatoportal vein [11, 13-15]. This hypothesis was initially generated more than a decade ago during experiments which showed that GLP-1 and the GLP-1 receptor trigger the vagus nerve activity in response to glucose [16-18] and subsequently confirmed by independent researchers [19-21], also in humans [22]. This pathway might play an important role in the neural control of glucagon secretion [23-26]. We recently further characterized this gut-to-brain-to-pancreas axis by using low dose oral DPP4i which ensured an increased portal vein GLP-1 concentration [15]. This procedure induced increased vagal firing rates, leading to the stimulation of insulin secretion without increasing systemic concentration of active GLP-1 [15]. Importantly, this regulation would be expected to be impaired in patients with autonomic neuropathy (AN). To investigate this hypothesis further and to avoid the interfering effect of glucose-induced insulin secretion on the regulation glucagon secretion, we subjected T1DM patients characterized by the presence or absence of autonomic diabetic neuropathy to a meal test without or with administration of 50 mg of vildagliptin 30 min before a meal test. Plasma glucose, glucagon, incretins, and pancreatic polypeptide (PP) and selected metabolite concentrations were assessed during the following 180 min.
| Patients and Methods | ▴Top |
Patients
We included 18 male and female patients with C-peptide negative T1DM, aged 18 - 75 years, insulin treated with multiple daily insulin injections (MDI) or continuous subcutaneous insulin infusion (CSII), diagnosed, according to the current guidelines [27], with confirmed (AN+) or without (AN-) cardiac autonomic neuropathy during autonomic testing realized within the year preceding the enrolment. HbA1c was ≤ 10% and stable (±1%). Females were required to be postmenopausal, surgically sterile, or using a reliable method of contraception. According to the hypothesis, the glucagon responses would be expected to differ markedly according to the presence or absence of neuropathy; therefore, inclusion of 18 individuals should provide the study with sufficient power. It is noteworthy that the study should still be considered only to generate the first evidences to support the hypothesis.
Main exclusion criteria were heart disease (NYHA class III or IV), renal failure (MDRD < 50 mL/min), proliferative retinopathy, liver disease, digestive disease, symptoms of gastroparesis (nausea, vomiting, epigastric pain, and early satiety), any medication that could interfere with the absorption of vildagliptin and ongoing corticosteroid therapy. Patients were allocated into two groups regarding their autonomic neuropathic status (AN- vs. AN+) that were matched for sex, age, and HbA1c, although diabetes duration and daily insulin needs were slightly higher in AN+ than in AN- patients. The characteristics of the 18 patients who completed the study are presented in Table 1.
 Click to view | Table 1. Baseline Characteristics of Patients |
Study design
This was a mechanistic study designed to evaluate whether an impaired autonomic nervous system (neuropathy) would hamper the glucagonostatic effect of the DPP4i, vildagliptin. Patients underwent a meal test (Fortimel Lactose Free containing 29 g carbohydrate, 20 g protein and 7 g lipids) without insulin bolus administration. This procedure ensures a stimulation of glucagon secretion. Patients were overnight fasted while receiving long acting insulin treatment (MDI patients) or basal rate continuous insulin infusion (CSII patients) without insulin bolus administration in the 3 h preceding the test. In order to avoid major postprandial hyperglycemia, patients had to reach a 0.7 - 1.80 g/L capillary glucose target prior to the meal test. No more than 28 days later, patients underwent the same procedure, except for a single 50 mg vildagliptin oral administration 30 min before the meal test.
Blood samples (tubes containing EDTA 100 nM, aprotinin 2.5 pM and diprotin A 0.1 mM) were obtained at 0, 15, 30, 60, 90, 120 and 180 min. The tubes were immediately centrifuged for 15 min at 4,000 rpm at 4 °C. Plasma was collected and aliquots stored at -80 °C until the concentrations of glucagon, total GIP, active and total GLP-1, PP, insulin, glucose, free fatty acids (FFAs), triglycerides, HDL, LDL, total cholesterol and lactate were assessed. At the end of the test, induced hyperglycemia was corrected under control of the investigator and patient remained in observation for 4 h to monitor unexpected adverse events. The protocol was reviewed and approved by Local Ethic Committee (CPP Sud Ouest and Outre Mer 1) and the French Competent Authority (ANSM). Each patient gave written informed consent prior to participation. This study has been registered on clinicaltrials.gov website, number NCT01452113.
Biochemical analyses
The concentration of plasma glucose was determined by an enzymatic colorimetric procedure (ABX Pentra Glucose HK CP). Plasma insulin concentrations were determined using the human ultrasensitive insulin ELISA kit (Mercodia, Uppsala, Sweden). PP concentration was determined using the human Pancreatic Polypeptide ELISA kit (Millipore, Billerica, USA). The concentrations of plasma FFAs and lactic acid were assessed by colorimetric assays. Lactate was assessed using lactate oxidase which triggers the release of hydrogen peroxide, which reacts with 4-aminoantipyrine and ESPAS, forming a colored complex in the presence of peroxidase. The intensity of the coloring is proportional to the amount of lactate present in the sample (ABX Pentra Glucose HK CP). FFAs were assessed by enzymatic colorimetric procedure according to the recommendation from the supplier (NEFA C, WAKO, Neus, DE).
Total GLP-1 concentrations were determined with RIA, using a C-terminally directed antiserum (code no. 89390) whereas the active form of the peptide was assessed using a highly specific sandwich ELISA with a sensitivity below 1 pmol/L as described in detail [28]. Total GIP was analyzed using a C-terminally directed antiserum (code no. 80867). Glucagon concentrations in plasma were measured after extraction of plasma with 70% ethanol (vol/vol, final concentration). The antibody employed (code no. 4305) is directed against the C-terminus of the glucagon molecule and therefore mainly measures glucagon of pancreatic origin [29, 30]. Standards were human glucagon and tracer was monoiodinated human glucagon (both gifts from NovoNordisk, Bagsvaerd). Sensitivity and detection limit is below 1 pmol/L, intra-assay coefficient of variation is below 6% at 20 - 30 pmol/L, and recovery of standard, added to plasma before extraction, is about 100% when corrected for losses inherent in the plasma extraction procedure.
LDL and HDL cholesterol were assessed by enzymatic colorimetric procedure (ABX Pentra LDL Direct CP and ABX Pentra HDL Direct CP respectively), total cholesterol was analyzed by enzymatic photometric procedure (CHOD-PAP from ABX Pentra Cholesterol CP), and triglycerides (TG) was assessed by an enzymatic colorimetric procedure (ABX Pentra Triglycerides CP).
Plasma DPP4 activity was assessed as previously described [15]. Briefly, DPP4 activity was assessed in 50 µL of plasma incubated with kit reagents for 2 h at 37 °C according to the manufacturers’ recommendations using recombinant DPP4 as a standard, expressed in nanograms per milliliter (DPP4 Glo protease assay; Promega, Madison, WI).
Autonomic diabetic neuropathy Ewing score
AN detection was based on Ewing battery (score ranging from 0 to 5) including at least two abnormal heart rate responses in cardio-vagal tests including deep breathing (6 cycles/min),Valsalva maneuver, 30/15 ratio independently of the presence or not of orthostatic hypotension, changes in blood pressure during standing. Orthostatic hypotension was defined as a reduction in systolic BP of at least 20 mm Hg or diastolic BP at least 10 mm Hg within 3 min standing test (AN+) or without (≤ 1 borderline response to cardio-vagal tests and no orthostatic hypotension) (AN-) cardiac autonomic neuropathy during autonomic testing realized within the year preceding the enrolment. The response to each test was considered as normal (0), borderline (0.5) or impaired (1) according to laboratory normal values for age [31]. According to Ewing score, patients were separated in two groups without (AN-, score < 0.5) or with (AN+, score > 2) confirmed autonomic failure.
Statistical analyses
Unless stated otherwise, quantitative variables are expressed as means ± SD and qualitative variables as number (percentage). Statistical analysis was performed using GraphPad Prism statistical software. In bivariate analysis, Chi-square test or Fischer’s exact test when appropriate was used to compare the distribution of qualitative variables between the groups. Means of quantitative variables were compared by Student’s t-test or Mann-Whitney test when appropriate. P < 0.05 was considered significant.
| Results | ▴Top |
Baseline characteristics of the patients
At baseline, patients with or without AN were matched for sex, age, insulin treatment, HbA1c, BMI, and all other major diabetic features (Table 1). It is noteworthy that the diabetes duration was slightly higher in the AN+ than in the AN- group.
Effect of autonomic diabetic neuropathy on plasma peptide and metabolite concentration profiles following a meal test without DPP4i
We first evaluated the impact of AN on the levels of circulating peptides (PP, glucagon, GLP-1 and GIP) and metabolites (glucose, lactates, FFA and lipids) following a meal test in T1DM patients. The plasma concentration of the PP rapidly increased after the first 15 min following the meal test, but to a lower extent in AN+ patients when compared to AN- controls (Fig. 1A). The plasma glucagon concentrations increased to the same extent in both groups of patients within the first 30 min after the meal test. However, they remained elevated only in the AN+ group of patients while returning towards basal values in AN- patients (Fig. 1B) showing an altered regulation of glucagon in the presence of AN. We then evaluated whether it was linked to an impaired secretion of GLP-1. The plasma concentration of active GLP-1 increased in both groups of patients although to a much lower extent in AN+ patients (Fig. 1C) whereas the plasma concentration profiles of total GLP-1 were similar in both groups (not shown). Thus, the molar ratio of glucagon to active GLP-1 was higher in the AN+ patients than in AN- patients. The plasma GIP concentration profiles were similar in both groups (Fig. 1D).
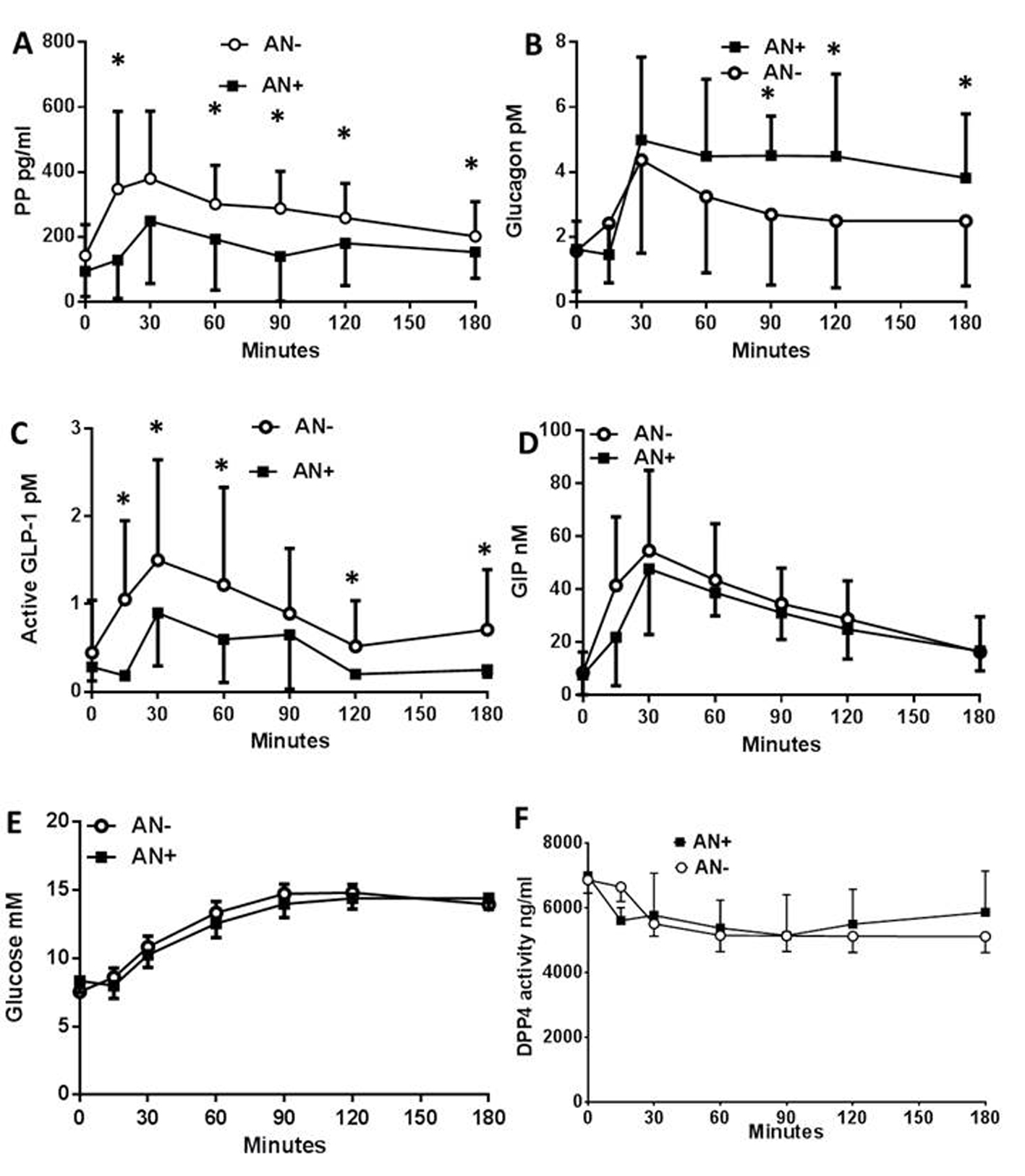 Click for large image | Figure 1. Plasma peptide and metabolite concentrations in type 1 diabetic patients with or without autonomic neuropathy following a meal test. Plasma concentration of pancreatic polypeptide (A), glucagon (B), active GLP-1 (C), total GIP (D), glucose (E), and plasma DPP4 activity (F), following the meal test in type 1 diabetic patients with autonomic neuropathy (AN+, black square) or without (AN-, open rings). Data are presented in means ± SD. *Difference statistically significant when P < 0.05. |
We then assessed the changes in plasma metabolites. The plasma glucose (Fig. 1E), plasma lactate, FFA, TG, HDL, LDL and cholesterol concentrations were also similar between both groups. The plasma DPP4 activity was similar in the two groups showing a 15-20% decrease during the first 15 - 30 min following the meal test which was maintained for the rest of the observation period (Fig. 1F). There were no differences in the circulating exogenous insulin concentration (not shown). All patients were C-peptide negative, therefore, endogenous insulin did not contribute.
Effect of autonomic diabetic neuropathy on the control by vildagliptin of plasma peptide and metabolite concentration profiles
The impact of neuropathy on the control of the concentration profiles of plasma peptides and metabolites in response to a DPP4i was then assessed in the same patients following another meal test less than 28 days later, and a single administration of the inhibitor. The DPP4i did not modify the plasma PP concentration profiles in patients with or without neuropathy (not shown) which remained therefore, similar to what observed in Figure 1A. In contrast, the DPP4i treatment significantly reduced the plasma concentrations of glucagon in AN- patients (Fig. 2A) whereas, this effect was lost for the AN+ patients since the plasma glucagon concentrations remained steadily elevated following the meal test (Fig. 2B). In both AN- and AN+ patients, the plasma concentrations of active GLP-1 were, as expected, increased by the DPP4i treatment (Fig. 2C, D). All other parameters, including glycemia, lactate, FFA, TG, and cholesterol remained unaffected by the acute DPP4i treatment irrespective of the neuropathy (not shown). The lack of metabolic impact of the single administration of DPP4i might be related to the need, in T1DM patients, of either insulin secretion or to a longer exposure to the treatment.
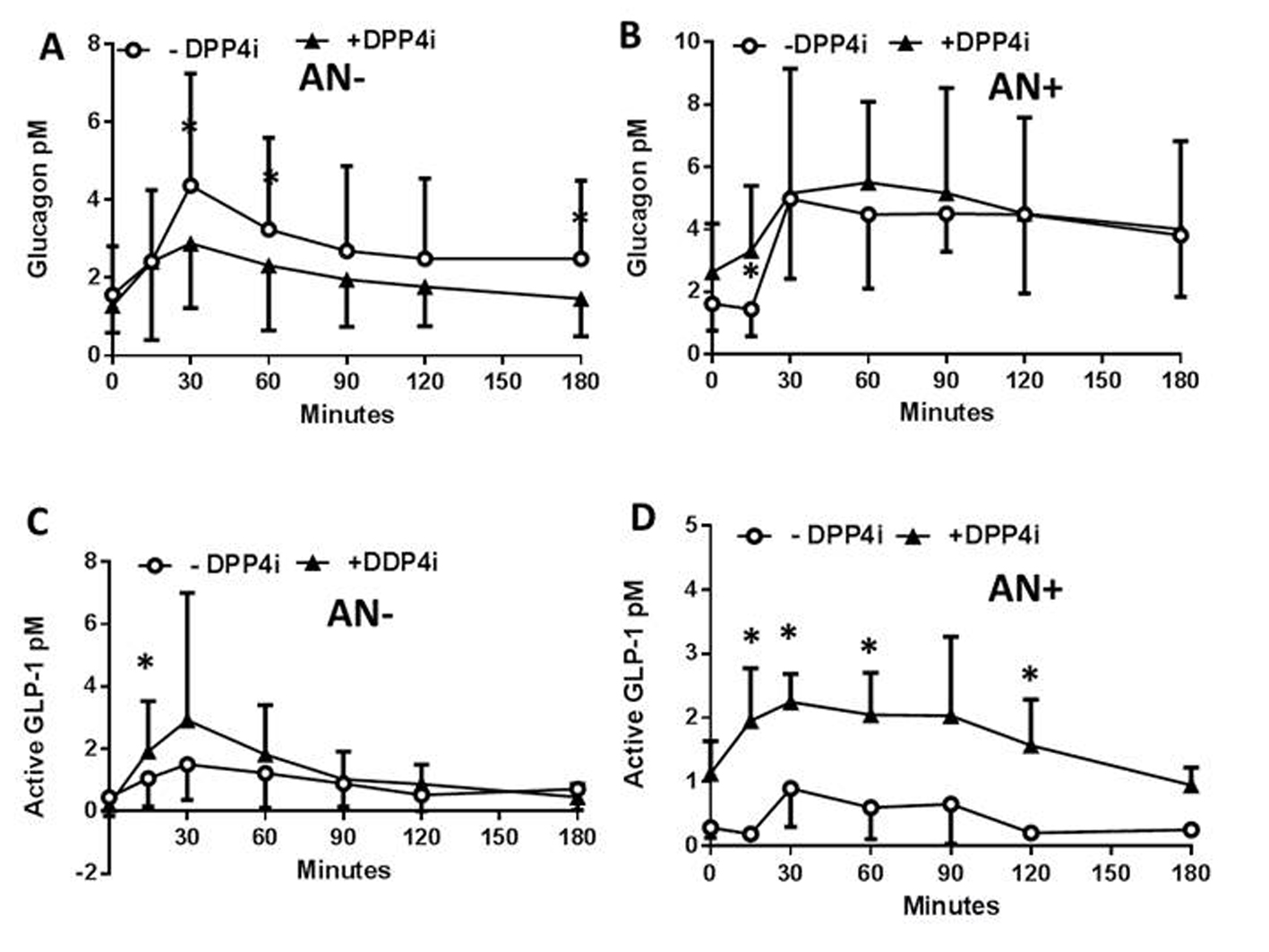 Click for large image | Figure 2. Plasma peptide and metabolite concentrations in type 1 diabetic patients with or without autonomic neuropathy following a single administration of vildagliptin and a meal test. In patients without (AN-) or with (AN+) autonomic neuropathy and following or not a single administration of a DPP4i vildagliptin (black triangles, and open rings, respectively) the plasma concentration of glucagon (A, B), and active GLP-1 (C, D), and the glucagon/active GLP-1 ratio were assessed. Data are presented in means ± SD. *Difference statistically significant when P < 0.05. |
| Discussion | ▴Top |
We here show in T1DM patients that plasma glucagon concentrations following a meal test may be lowered by a DPP4i, obviously via an insulin-independent mechanism, whereas a similar inhibition by DPP4i is not seen in patients with AN indicating that the autonomic nervous system activity is involved in inhibitory activity of the DPP4i. DPP4 inhibition increased similarly the levels of intact GLP-1 in patients with as well as without neuropathy. Therefore, given that elevated levels of intact GLP-1 are responsible for the normal inhibition of glucagon secretion, our data also suggest that AN induces a state of glucagon unresponsiveness representing a form of GLP-1 resistance.
Patients with both T1DM and T2DM have impaired regulation of glucagon secretion which may be responsible for excessive hepatic glucose production [32, 33]. In addition, unlike healthy humans, patients with T1DM do not respond to hypoglycemia with increased glucagon secretion, and this is thought to be of importance for the high frequency of hypoglycemic attacks during insulin therapy in these patients. The mechanisms involved are yet unclear but could be related, to some degree, to AN which often affects these patients [34, 35]. We provide here evidence to support this hypothesis by showing that AN is associated with an exaggerated glucagon response to a meal test in insulin-deprived T1DM patients. The mechanism could not be related to disturbed insulin action since all patients, with or without neuropathy, were C-peptide negative. These observations may therefore be consistent with a role for the autonomic nervous system in the control of glucagon secretion [36, 37]. Previous work in rodents showed that vagotomy [38] as well as ventromedial hypothalamic lesions [39] hampers the regulation of glucagon secretion following feeding in rats. We here further showed that the meal responses of PP were lower in patients with neuropathy in agreement with the notion that activity in efferent cholinergic neurons of the parasympathetic nervous system is responsible for a considerable part of PP response to a meal. Acetylcholine, the parasympathetic transmitter has been shown to reduce glucagon secretion on isolated perfused pancreas [40]. The exaggerated glucagon response in the neuropathy patients might therefore be due to an impaired cholinergic, parasympathetic inhibition of glucagon secretion. A 80% reduction in the number of nerve endings in direct contact with alpha-cells was noted type 1 diabetes BB rats [41] which could be responsible for an impaired glucagon regulation as well as reduced PP secretion. However, acetylcholine activates glucagon secretion from isolated pancreatic alpha cells [42, 43], indicating that glucagonostatic effect of acetylcholine on the whole pancreas is due to a more complex mechanism, perhaps involving inhibition of somatostatin secretion [37, 44]. Our data are in agreement with this conclusion since the plasma glucagon to PP concentration ratio increased during the meal test in AN+ patients while it remained low and steady under control in AN- patients. Furthermore, the glucose unresponsiveness of the patients with AN could be due to a state of GLP-1 resistance. The impaired glucagonostatic action of the active GLP-1 was not due to an impaired secretion of the peptide since total plasma GLP-1 concentration was normal. Hence, a tight relationship is suggested between the autonomic parasympathetic nerve activity and the action of endogenous GLP-1 on glucagon concentration. Our conclusion is also supported by recent data from the literature showing that the deletion of the GLP-1 receptor specifically in the β cells did not impair oral glucose induced insulin secretion in mice [45] further supporting the importance of GLP-1 receptors expressed in other than beta cells. Therefore, as we previously suggested, endogenous GLP-1 may control pancreatic endocrine secretions mainly through neural mechanisms [10, 46-48], whereas GLP-1 receptor agonists reaching high circulating concentrations of active agonist would control glycemia through a direct effect on β cells [49].
Interestingly, neuropathy also appeared to reduce meal test-induced active GLP-1 plasma concentration suggesting that GLP-1 homeostasis is under neural regulation. The impaired GLP-1 response may contribute to the impaired regulation of glucagon secretion observed in patients with AN. Possibly an impaired function of the autonomic nervous system may result in impaired GLP-1 secretion, perhaps by influencing gastrointestinal motility [50-52]. However, our data show that the total GLP-1 concentration was not dramatically affected by the AN, most likely ruling out impaired neural regulation of GLP-1 secretion. Furthermore, no change in DPP4 activity was observed. The acute changes observed in plasma glucagon and GLP-1 concentrations did not affect the overall glycemic profiles following the meal test suggesting that chronic long term rather than acute changes in plasma GLP-1 and glucagon concentrations are required to improve the glycemic control.
Since our data might suggest that lower concentrations of active GLP-1 could explain the exaggerated glucagon response in AN, we treated the AN+ T1DM patients with a DPP4i to enhance GLP-1 action. Indeed, the DPP4i efficiently increased the plasma concentrations of active GLP-1 but, however, failed to improve the glucagon concentration profiles during the meal test in AN+ patient. Our findings therefore suggest that AN increases meal-induced glucagon secretion through a mechanism preventing the glucagonostatic effect of enteric GLP-1 on the gut to alpha cell axis in T1DM patients.
In T1DM patients without AN, our data show that the administration of a DPP4i, to enhance the physiological action of gut-released GLP-1, inhibits glucagon secretion by an insulin-independent mechanism with the additional information that the mechanism requires an intact autonomic nervous system. Also GLP-1 agonists inhibit glucagon secretion via insulin-independent mechanisms [5, 53]. Our data are also in agreement with previous observations with vildagliptin in patients with T1DM [3, 54].
Although we cannot rule out that the GLP-1 receptor present at the surface of the alpha cells could be responsible for the direct glucagonostatic effect of the GLP-1 secreted by the gut, the demonstration that vagotomy abolishes it supports the role of a neural mechanism [22]. Electrophysiological recording of the vagus nerve discharge in the presence of GLP-1 demonstrates the direct effect of the peptide [16, 55]. Long vago-vagal reflexes in turn would regulate pancreatic and extrapancreatic functions such as insulin secretion [13], whole body glucose turnover [46, 56-59] and liver metabolism [60] participating in general gut-to-brain-to-periphery regulation of metabolism. Impaired gastric emptying in patients with AN could have contributed to the impaired glucagon secretion, but in our study patients with symptomatic gastroparesis were excluded, although gastric emptying was not measured in all the patients. Furthermore, the glycemic peak and the maximal early plasma glucagon concentrations reached following the meal test were similar in patients with or without neuropathy showing that the overall meal has been most likely similarly absorbed. A recent study in patients with T2DM demonstrated no effect of vildagliptin on the rate of gastric emptying or meal glucose appearance [61] suggesting that this issue was not at play in our study.
Importantly and to the best of our knowledge, the integrity of the autonomic nervous system has never been considered as a criterion to predict the responsiveness of alpha cells to the glucagonostatic effect of a GLP-1-based therapy. This criterion could define responders versus non-responder patients. In addition to DPP4i responsiveness, our conclusion could also be true for GLP-1 receptor agonists which would require an integrated autonomic nervous system to efficiently control glucagon secretion and hence glycemia. Although, whether high circulating GLP-1 concentration, as obtained using GLP-1 receptor agonist, is sufficient to activate the gut brain to pancreas axis remains to be determined.
Altogether, we here show that the AN, which characterizes many T1DM and T2DM patients [34, 35], impairs the gut to alpha cell axis regarding GLP-1 regulated glucagon secretion. Consequently, the autonomic diabetic neuropathy could hamper the regulation of glycemia by DPP4i. This would be of major importance for the treatment of T2DM since both insulin and glucagon secretion would be impaired by AN.
Acknowledgement
This work was supported by a grant from Novartis Pharma to Remy Burcelin and a grant of the University Hospital (CHU de Toulouse) to Emilie Lobinet. We would like to thank Sophie Legonidec from the phenotyping platform of US006/CREFRE (INSERM/UPS). RB has received honorarium from Novartis. SD is emplyoee of Novartis Pharma.
| References | ▴Top |
- Garg SK, Moser EG, Bode BW, Klaff LJ, Hiatt WR, Beatson C, Snell-Bergeon JK. Effect of sitagliptin on post-prandial glucagon and GLP-1 levels in patients with type 1 diabetes: investigator-initiated, double-blind, randomized, placebo-controlled trial. Endocr Pract. 2013;19(1):19-28.
doi pubmed - Ellis SL, Moser EG, Snell-Bergeon JK, Rodionova AS, Hazenfield RM, Garg SK. Effect of sitagliptin on glucose control in adult patients with Type 1 diabetes: a pilot, double-blind, randomized, crossover trial. Diabet Med. 2011;28(10):1176-1181.
doi pubmed - Foley JE, Ligueros-Saylan M, He YL, Holst JJ, Deacon CF, Dunning BE, Leone-Jones A, et al. Effect of vildagliptin on glucagon concentration during meals in patients with type 1 diabetes. Horm Metab Res. 2008;40(10):727-730.
doi pubmed - Creutzfeldt WO, Kleine N, Willms B, Orskov C, Holst JJ, Nauck MA. Glucagonostatic actions and reduction of fasting hyperglycemia by exogenous glucagon-like peptide I(7-36) amide in type I diabetic patients. Diabetes Care. 1996;19(6):580-586.
doi pubmed - Kielgast U, Holst JJ, Madsbad S. Antidiabetic actions of endogenous and exogenous GLP-1 in type 1 diabetic patients with and without residual beta-cell function. Diabetes. 2011;60(5):1599-1607.
doi pubmed - Liu M, Seino S, Kirchgessner AL. Identification and characterization of glucoresponsive neurons in the enteric nervous system. J Neurosci. 1999;19(23):10305-10317.
pubmed - Marty N, Dallaporta M, Foretz M, Emery M, Tarussio D, Bady I, Binnert C, et al. Regulation of glucagon secretion by glucose transporter type 2 (glut2) and astrocyte-dependent glucose sensors. J Clin Invest. 2005;115(12):3545-3553.
doi pubmed - Miki T, Liss B, Minami K, Shiuchi T, Saraya A, Kashima Y, Horiuchi M, et al. ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nat Neurosci. 2001;4(5):507-512.
pubmed - Seino S, Iwanaga T, Nagashima K, Miki T. Diverse roles of K(ATP) channels learned from Kir6.2 genetically engineered mice. Diabetes. 2000;49(3):311-318.
doi pubmed - Knauf C, Cani PD, Perrin C, Iglesias MA, Maury JF, Bernard E, Benhamed F, et al. Brain glucagon-like peptide-1 increases insulin secretion and muscle insulin resistance to favor hepatic glycogen storage. J Clin Invest. 2005;115(12):3554-3563.
doi pubmed - Hjollund KR, Deacon CF, Holst JJ. Dipeptidyl peptidase-4 inhibition increases portal concentrations of intact glucagon-like peptide-1 (GLP-1) to a greater extent than peripheral concentrations in anaesthetised pigs. Diabetologia. 2011;54(8):2206-2208.
doi pubmed - de Heer J, Rasmussen C, Coy DH, Holst JJ. Glucagon-like peptide-1, but not glucose-dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas. Diabetologia. 2008;51(12):2263-2270.
doi pubmed - Balkan B, Li X. Portal GLP-1 administration in rats augments the insulin response to glucose via neuronal mechanisms. Am J Physiol Regul Integr Comp Physiol. 2000;279(4):R1449-1454.
pubmed - Preitner F, Ibberson M, Franklin I, Binnert C, Pende M, Gjinovci A, Hansotia T, et al. Gluco-incretins control insulin secretion at multiple levels as revealed in mice lacking GLP-1 and GIP receptors. J Clin Invest. 2004;113(4):635-645.
doi pubmed - Waget A, Cabou C, Masseboeuf M, Cattan P, Armanet M, Karaca M, Castel J, et al. Physiological and pharmacological mechanisms through which the DPP-4 inhibitor sitagliptin regulates glycemia in mice. Endocrinology. 2011;152(8):3018-3029.
doi pubmed - Nakabayashi H, Nishizawa M, Nakagawa A, Takeda R, Niijima A. Vagal hepatopancreatic reflex effect evoked by intraportal appearance of tGLP-1. Am J Physiol. 1996;271(5 Pt 1):E808-813.
pubmed - Nishizawa M, Nakabayashi H, Kawai K, Ito T, Kawakami S, Nakagawa A, Niijima A, et al. The hepatic vagal reception of intraportal GLP-1 is via receptor different from the pancreatic GLP-1 receptor. J Auton Nerv Syst. 2000;80(1-2):14-21.
doi - Nakagawa A, Satake H, Nakabayashi H, Nishizawa M, Furuya K, Nakano S, Kigoshi T, et al. Receptor gene expression of glucagon-like peptide-1, but not glucose-dependent insulinotropic polypeptide, in rat nodose ganglion cells. Auton Neurosci. 2004;110(1):36-43.
doi pubmed - Nishizawa M, Nakabayashi H, Uehara K, Nakagawa A, Uchida K, Koya D. Intraportal GLP-1 stimulates insulin secretion predominantly through the hepatoportal-pancreatic vagal reflex pathways. Am J Physiol Endocrinol Metab. 2013;305(3):E376-387.
doi pubmed - Labouesse MA, Stadlbauer U, Weber E, Arnold M, Langhans W, Pacheco-Lopez G. Vagal afferents mediate early satiation and prevent flavour avoidance learning in response to intraperitoneally infused exendin-4. J Neuroendocrinol. 2012;24(12):1505-1516.
doi pubmed - Bucinskaite V, Tolessa T, Pedersen J, Rydqvist B, Zerihun L, Holst JJ, Hellstrom PM. Receptor-mediated activation of gastric vagal afferents by glucagon-like peptide-1 in the rat. Neurogastroenterol Motil. 2009;21(9):978-e978.
doi pubmed - Plamboeck A, Veedfald S, Deacon CF, Hartmann B, Wettergren A, Svendsen LB, Meisner S, et al. Characterisation of oral and i.v. glucose handling in truncally vagotomised subjects with pyloroplasty. Eur J Endocrinol. 2013;169(2):187-201.
doi pubmed - Gromada J, Franklin I, Wollheim CB. Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr Rev. 2007;28(1):84-116.
doi pubmed - Burcelin R, Thorens B. Evidence that extrapancreatic GLUT2-dependent glucose sensors control glucagon secretion. Diabetes. 2001;50(6):1282-1289.
doi pubmed - Taborsky GJ, Jr., Ahren B, Mundinger TO, Mei Q, Havel PJ. Autonomic mechanism and defects in the glucagon response to insulin-induced hypoglycaemia. Diabetes Nutr Metab. 2002;15(5):318-322; discussion 322-313.
- Burcelin R, Knauf C, Cani PD. Pancreatic alpha-cell dysfunction in diabetes. Diabetes Metab. 2008;34(Suppl 2):S49-55.
doi - Spallone V, Ziegler D, Freeman R, Bernardi L, Frontoni S, Pop-Busui R, Stevens M, et al. Cardiovascular autonomic neuropathy in diabetes: clinical impact, assessment, diagnosis, and management. Diabetes Metab Res Rev. 2011;27(7):639-653.
doi pubmed - Wewer Albrechtsen NJ, Bak MJ, Hartmann B, Christensen LW, Kuhre RE, Deacon CF, Holst JJ. Stability of glucagon-like peptide 1 and glucagon in human plasma. Endocr Connect. 2015;4(1):50-57.
doi pubmed - Holst JJ. Evidence that enteroglucagon (II) is identical with the C-terminal sequence (residues 33-69) of glicentin. Biochem J. 1982;207(3):381-388.
doi pubmed - Wewer Albrechtsen NJ, Hartmann B, Veedfald S, Windelov JA, Plamboeck A, Bojsen-Moller KN, Idorn T, et al. Hyperglucagonaemia analysed by glucagon sandwich ELISA: nonspecific interference or truly elevated levels? Diabetologia. 2014;57(9):1919-1926.
doi pubmed - Pavy-Le Traon A, Fontaine S, Tap G, Guidolin B, Senard JM, Hanaire H. Cardiovascular autonomic neuropathy and other complications in type 1 diabetes. Clin Auton Res. 2010;20(3):153-160.
doi pubmed - Unger RH, Orci L. The role of glucagon in diabetes. Compr Ther. 1982;8(1):53-59.
pubmed - Unger RH, Dobbs RE, Orci L. Insulin, glucagon, and somatostatin secretion in the regulation of metabolism. Annu Rev Physiol. 1978;40:307-343.
doi pubmed - Ewing DJ, Clarke BF. Autonomic neuropathy: its diagnosis and prognosis. Clin Endocrinol Metab. 1986;15(4):855-888.
doi - Ziegler D. Diabetic cardiovascular autonomic neuropathy: prognosis, diagnosis and treatment. Diabetes Metab Rev. 1994;10(4):339-383.
doi pubmed - Holst JJ, Gronholt R, Schaffalitzky de Muckadell OB, Fahrenkrug J. Nervous control of pancreatic endocrine secretion in pigs. II. The effect of pharmacological blocking agents on the response to vagal stimulation. Acta Physiol Scand. 1981;111(1):9-14.
doi pubmed - Holst JJ, Schwartz TW, Knuhtsen S, Jensen SL, Nielsen OV. Autonomic nervous control of the endocrine secretion from the isolated, perfused pig pancreas. J Auton Nerv Syst. 1986;17(1):71-84.
doi - Martin JR, Novin D, Vanderweele DA. Loss of glucagon suppression of feeding after vagotomy in rats. Am J Physiol. 1978;234(3):E314-318.
pubmed - Le Magnen J. Metabolic and feeding patterns: role of sympathetic and parasympathetic efferent pathways. J Auton Nerv Syst. 1984;10(3-4):325-335.
doi - Holst JJ, Schaffalitzky de Muckadell OB, Fahrenkrug J, Lindkaer S, Nielsen OV, Schwartz TW. Nervous control of pancreatic endocrine secretion in pigs. III. The effect of acetylcholine on the pancreatic secretion of insulin and glucagon. Acta Physiol Scand. 1981;111(1):15-22.
doi pubmed - Tominaga M, Maruyama H, Vasko MR, Baetens D, Orci L, Unger RH. Morphologic and functional changes in sympathetic nerve relationships with pancreatic alpha-cells after destruction of beta-cells in rats. Diabetes. 1987;36(3):365-373.
doi pubmed - Adeghate E, Ponery AS, Pallot DJ, Singh J. Distribution of neurotransmitters and their effects on glucagon secretion from the in vitro normal and diabetic pancreatic tissues. Tissue Cell. 2000;32(3):266-274.
doi pubmed - Funakoshi A, Yasunami Y, Ryu S, Shinozaki H, Jimi A, Ikeda S. Acetylcholine regulates glucagon secretion from human glucagonoma cells. J Gastroenterol. 1994;29(6):797-799.
doi pubmed - Holst JJ, Jensen SL, Knuhtsen S, Nielsen OV. Autonomic nervous control of pancreatic somatostatin secretion. Am J Physiol. 1983;245(6):E542-548.
pubmed - Smith EP, An Z, Wagner C, Lewis AG, Cohen EB, Li B, Mahbod P, et al. The role of beta cell glucagon-like peptide-1 signaling in glucose regulation and response to diabetes drugs. Cell Metab. 2014;19(6):1050-1057.
doi pubmed - Cabou C, Burcelin R. GLP-1, the gut-brain, and brain-periphery axes. Rev Diabet Stud. 2011;8(3):418-431.
doi pubmed - Holst JJ, Burcelin R, Nathanson E. Neuroprotective properties of GLP-1: theoretical and practical applications. Curr Med Res Opin. 2011;27(3):547-558.
doi pubmed - Burcelin R. The gut-brain axis: a major glucoregulatory player. Diabetes Metab. 2010;36(Suppl 3):S54-58.
doi - Sisley S, Gutierrez-Aguilar R, Scott M, D'Alessio DA, Sandoval DA, Seeley RJ. Neuronal GLP1R mediates liraglutide's anorectic but not glucose-lowering effect. J Clin Invest. 2014;124(6):2456-2463.
doi pubmed - Anini Y, Hansotia T, Brubaker PL. Muscarinic receptors control postprandial release of glucagon-like peptide-1: in vivo and in vitro studies in rats. Endocrinology. 2002;143(6):2420-2426.
doi pubmed - Hansen L, Lampert S, Mineo H, Holst JJ. Neural regulation of glucagon-like peptide-1 secretion in pigs. Am J Physiol Endocrinol Metab. 2004;287(5):E939-947.
doi pubmed - Anini Y, Brubaker PL. Muscarinic receptors control glucagon-like peptide 1 secretion by human endocrine L cells. Endocrinology. 2003;144(7):3244-3250.
doi pubmed - Kielgast U, Asmar M, Madsbad S, Holst JJ. Effect of glucagon-like peptide-1 on alpha- and beta-cell function in C-peptide-negative type 1 diabetic patients. J Clin Endocrinol Metab. 2010;95(5):2492-2496.
doi pubmed - Farngren J, Persson M, Schweizer A, Foley JE, Ahren B. Vildagliptin reduces glucagon during hyperglycemia and sustains glucagon counterregulation during hypoglycemia in type 1 diabetes. J Clin Endocrinol Metab. 2012;97(10):3799-3806.
doi pubmed - Nishizawa M, Nakabayashi H, Uchida K, Nakagawa A, Niijima A. The hepatic vagal nerve is receptive to incretin hormone glucagon-like peptide-1, but not to glucose-dependent insulinotropic polypeptide, in the portal vein. J Auton Nerv Syst. 1996;61(2):149-154.
doi - Burcelin R, Da Costa A, Drucker D, Thorens B. Glucose competence of the hepatoportal vein sensor requires the presence of an activated glucagon-like peptide-1 receptor. Diabetes. 2001;50(8):1720-1728.
doi pubmed - Johnson KM, Edgerton DS, Rodewald T, Scott M, Farmer B, Neal D, Cherrington AD. Intraportally delivered GLP-1, in the presence of hyperglycemia induced via peripheral glucose infusion, does not change whole body glucose utilization. Am J Physiol Endocrinol Metab. 2008;294(2):E380-384.
doi pubmed - Johnson KM, Edgerton DS, Rodewald T, Scott M, Farmer B, Neal D, Cherrington AD. Intraportal GLP-1 infusion increases nonhepatic glucose utilization without changing pancreatic hormone levels. Am J Physiol Endocrinol Metab. 2007;293(4):E1085-1091.
doi pubmed - Nishizawa M, Moore MC, Shiota M, Gustavson SM, Snead WL, Neal DW, Cherrington AD. Effect of intraportal glucagon-like peptide-1 on glucose metabolism in conscious dogs. Am J Physiol Endocrinol Metab. 2003;284(5):E1027-1036.
doi pubmed - Dardevet D, Moore MC, Neal D, DiCostanzo CA, Snead W, Cherrington AD. Insulin-independent effects of GLP-1 on canine liver glucose metabolism: duration of infusion and involvement of hepatoportal region. Am J Physiol Endocrinol Metab. 2004;287(1):E75-81.
doi pubmed - Vella A, Bock G, Giesler PD, Burton DB, Serra DB, Saylan ML, Deacon CF, et al. The effect of dipeptidyl peptidase-4 inhibition on gastric volume, satiation and enteroendocrine secretion in type 2 diabetes: a double-blind, placebo-controlled crossover study. Clin Endocrinol (Oxf). 2008;69(5):737-744.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.