

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website http://www.jofem.org |
Case Report
Volume 1, Number 2, June 2011, pages 87-89
Paucity of ACTH and Androgen Receptors in Massive Myelolipoma of Congenital Adrenal Hyperplasia Patient: More Insight Into Pathogenesis
Lai Y Wonga, Peter Campbellb, Takeshi Usuic, Anthony J O’Sullivana, d
aDepartments of Medicine and Endocrinology, St. George Hospital and the University of New South Wales, Australia
bDepartment of Breast and Endocrine Surgery, St. George Hospital, Sydney, Australia
cClinical Research Institute, National Hospital Organization Kyoto Medical Center, Japan
dCorresponding author: Anthony J O’Sullivan, Department of Medicine, St. George Hospital, Kogarah, NSW 2217, Australia
Manuscript accepted for publication May 26, 2011
Short title: Myelolipoma and Congenital Adrenal Hyperplasia
doi: https://doi.org/10.4021/jem22w
| Abstract | ▴Top |
Adrenal myelolipomas in congenital adrenal hyperplasia (CAH) are rare. The pathogenesis is unknown but adrenocorticotropic hormone (ACTH) is implicated to play a key role. Demonstration of paucity of ACTH and androgen receptors in these tumors may suggest otherwise. We describe a 54-year-old woman, presenting with bilateral massive myelolipoma associated with 21-hydroxylase deficiency CAH. Bilateral adrenalectomy was performed due to significant mass effect. Reverse transcriptase-polymerase chain reaction performed on tumor tissue showed no expression of the ACTH (MC2R - melanocortin 2 receptor) and androgen receptors. There are few published reports on pathogenesis of myelolipoma or in association with CAH. Recent evidence of chromosomal abnormality similar to hematopoietic cancer and inactivation of X chromosome in some myelolipomas suggestive of clonal proliferation from a common pluripotent stem cell has been found. However, origin of the hematopoietic tissue and the triggering factor for proliferation and development of the myelolipomas remain unclear. Nevertheless, the implication of these findings is exciting, as it suggests that development and proliferation of myelolipoma is not necessarily attributable to excess ACTH stimulation alone. Presence of local and systemic growth factors is possibly yet to be determined.
Keywords: Adrenal; Myelolipoma; Pathogenesis
| Introduction | ▴Top |
Adrenal masses in congenital adrenal hyperplasia (CAH) are common although there are only 20 published reports of adrenal myelolipomas in CAH. Myelolipomas are benign non-functioning tumors, consisting of adrenal, adipose and hematopoietic tissues. However, they can cause significant mass effect as their growth can potentially reach 6 kg in weight. The pathogenesis is unknown but in CAH patients, adrenocorticotropic hormone (ACTH) is implicated to play a key role. Demonstration of paucity of ACTH and androgen receptors in these tumors may suggest otherwise.
| Case Report | ▴Top |
We report a case of bilateral massive myelolipoma in a 21-hydroxylase deficiency CAH patient, with undetectable ACTH and androgen receptors in the tumor. The patient was a 54-year-old woman who initially presented to us 9 years ago for review of CAH. Diagnosis of CAH was made at birth, and corticosteroid replacement was commenced. She was poorly compliant and had weaned herself off corticosteroids by the age of 18.
She suffered primary amenorrhea and developed significant hirsutism and androgenization after puberty. Enlarged adrenal glands were discovered incidentally on CT scan at age 26 following a motor vehicle accident. Adrenal carcinoma was considered but surgery was refused by the patient on the basis that she was asymptomatic. She was subsequently lost to follow up.
She had been well until age 46 when she developed severe nausea, abdominal discomfort and vomiting which settled with supportive treatment. Corticosteroid treatment was recommenced. We first saw her the following year in 2001. Unfortunately, she suffered significant adverse corticosteroid effects with weight gain, lethargy and mood lability.
Clinically, she was androgenized with a male distribution of facial and chest hair. There was significant breast atrophy. Abdominal examination revealed an ill-defined mass on the left. She had evidence of genital androgenization. Her blood results on corticosteroid treatment showed suppressed ACTH, elevated 17-OH progesterone 31 nmol/L (normal range < 10 nmol/L), and testosterone 4.3 nmol/L (normal range 1.7 - 3.5 nmol/L). Her androstenedione, DHEAS and electrolytes levels were normal. She was noncompliant and chose to cease her corticosteroid due to adverse effects. Follow-up of her hormonal assays off corticosteroid showed markedly elevated ACTH 530 nmol/L (normal range < 12.1 pmol/L), 17-OH progesterone 795 nmol/L, testosterone 14.4 nmol/L, androstenedione 59.2 nmol/L (normal range 1.4 - 8.1 nmol/L), cortisol random sample 110 nmol/L (normal range 100 - 690 nmol/L) and normal electrolytes. Genetic studies confirmed compound heterozygous CYP21A2 mutation, with c328-335 deletion (salt wasting phenotype) and c293-13A/C>G mutation (salt wasting or simple virilizing phenotype) detected consistent with simple virilizing phenotype determined by the allele with the least severe phenotype.
Between 2001 and 2007, she suffered from progressively increasing left sided abdominal pain radiating to the flank requiring multiple admissions for pain relief. Serial CT scans showed a substantial increase in size of both her adrenal glands as a cause for her symptoms, with compression of the inferior vena cava and 30% reduced function in the left kidney.
Bilateral open adrenalectomy with perioperative corticosteroid cover was performed in view of above findings and her recovery was uneventful. Postoperatively, she was commenced on prednisolone and fludrocortisone. The right adrenal gland weighed 308 g (measuring 11.5 × 10 × 6 cm) and left adrenal gland weighed 5 kg (measuring 24 × 23.5 × 9 cm). Macroscopically the tumors consisted of dark tan globular tissue surrounded by fatty tissue with areas of hemorrhage. Microscopically the tumors were dominated by mature adipose tissue with some mild fibrosis and myxoid change with extensive infiltration by hemopoietic tissue including all three cell lineages (Fig. 1). In addition, adrenal cortical tissue and focally adrenal medullary tissue were identified, consistent with adrenal cortical hyperplasia and myelolipoma. The patient consented to genetic analysis of the tumor. Specimens were sent for RT-PCR (reverse transcription-polymerase chain reaction) in Japan, and no expression of the ACTH (MC2R - melanocortin 2 receptor) and androgen receptors was detected (Fig. 2).
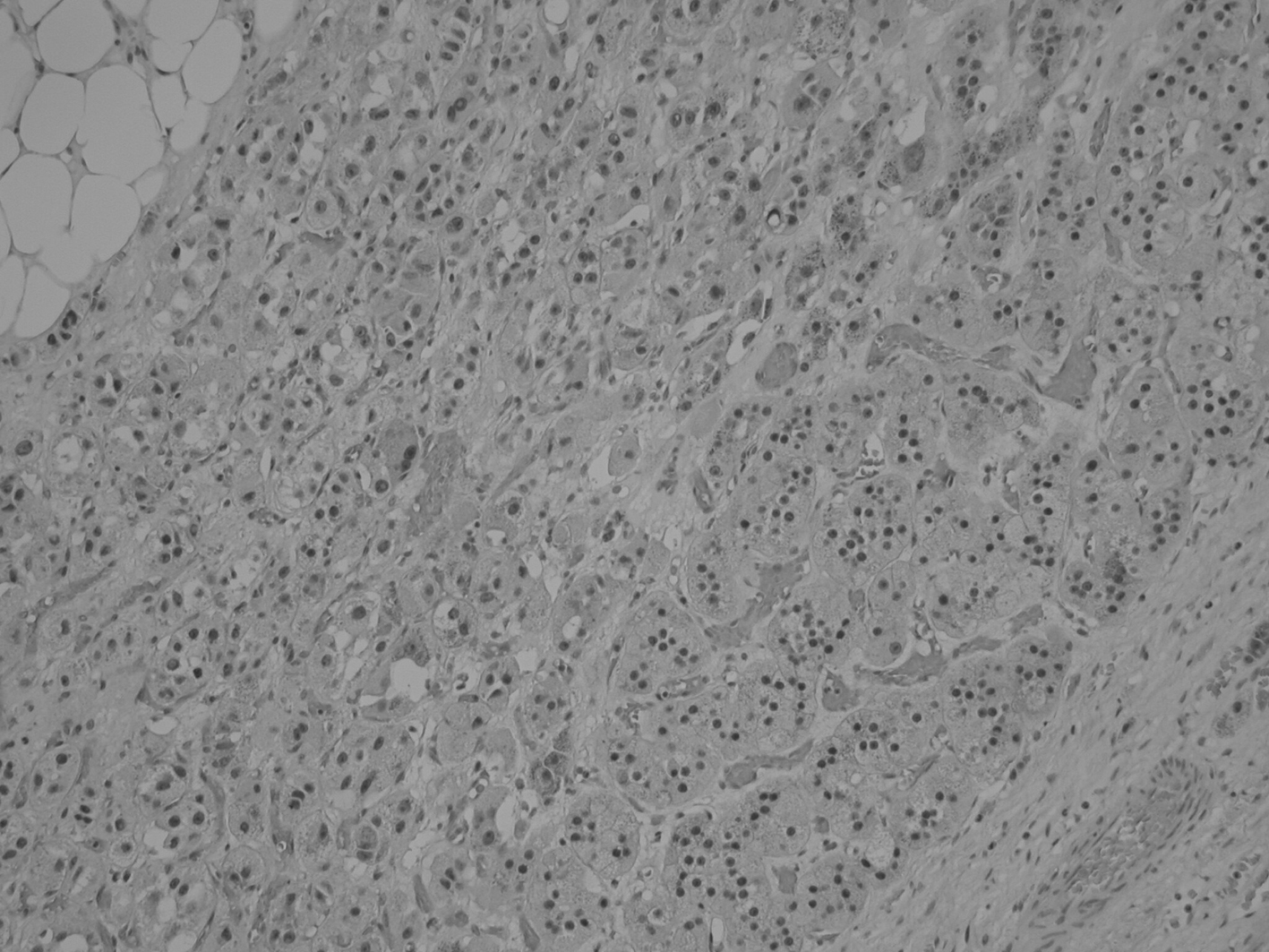 Click for large image | Figure 1. Histology of the adrenal myelolipoma demonstrating presence of adipose, hematopoietic and adrenal cortical cells. |
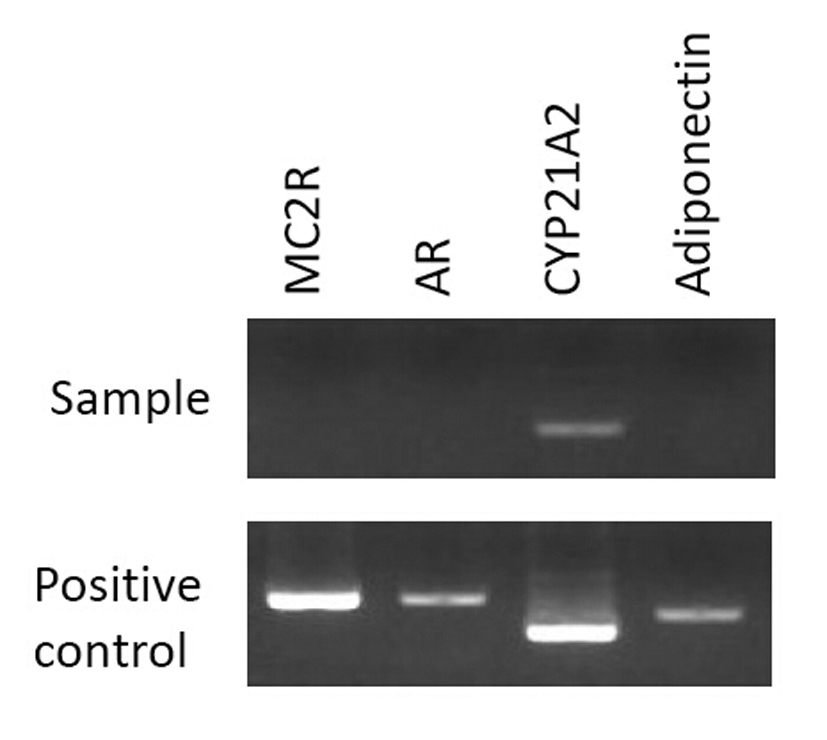 Click for large image | Figure 2. Analysis by RT-PCR for MC2R, AR, CYP21A2, and adiponectin. No expression was detected for MC2R and AR in the sample. Positive control cDNAs were obtained from the previously reported samples [5]. |
| Discussion | ▴Top |
Available published reports on pathogenesis of myelolipoma or in association with CAH patients are scarce. Recent evidence of chromosomal abnormality similar to hematopoietic cancer [1] and inactivation of X chromosome in some myelolipomas indicative of clonal proliferation from a common pluripotent stem cell has been found [2].
The origin of the hematopoietic tissue is unclear but existing theories include presence of embryonic bone marrow rests in the adrenal tissue, embolization of viable bone marrow to the adrenals, and adrenocortical cell metaplasia from the reticuloendothelial cells in capillaries. However, adipose and myeloid cells originating from progenitor cells in the adrenal stroma seems to be the most likely explanation [3].
There are sufficient data to suggest ACTH as a stimulant for the proliferation of myeloid and adipose tissues. Notably, myelolipomas are prevalent in patients with excessive ACTH production, especially in congenital adrenal hyperplasia, Nelson’s syndrome, and Addison’s disease. Myeloid metaplasia in the adrenal cortex has been demonstrated in severely burnt patients, and cancer patients who were under intense stress [4].
However, Hagiwara and Usui et al [5], in the first published report using RT-PCR on a patient with massive myelolipoma, failed to identify gene expression for the main ACTH receptors consisting of MC2R (melanocortin 2 receptor) and MC5R (melanocortin 5 receptor), as well as androgen receptors (AR). In view of the massive size of tumor, they postulated these receptors could still have been overexpressed in the early stage of its development. There may also be an increased sensitivity to or paracrine stimulus from either ACTH or androgen. Similar results were obtained in our patient’s specimen.
There are limitations to the specimen analyzed. The results may not be unequivocal, as only a small portion of a massive tumor was tested. Nevertheless, the implication of these findings is exciting, as the above data suggest that development and proliferation of myelolipoma is not necessarily attributable to excess ACTH stimulation alone. Presence of local and systemic growth factors is possibly yet to be determined.
| References | ▴Top |
- Chang KC, Chen PI, Huang ZH, Lin YM, Kuo PL. Adrenal myelolipoma with translocation (3;21)(q25;p11). Cancer Genet Cytogenet 2002;134(1):77-80.
pubmed doi - Bishop E, Eble JN, Cheng L, Wang M, Chase DR, Orazi A, O'Malley DP. Adrenal myelolipomas show nonrandom X-chromosome inactivation in hematopoietic elements and fat: support for a clonal origin of myelolipomas. Am J Surg Pathol 2006;30(7):838-843.
pubmed doi - Han M, Burnett AL, Fishman EK, Marshall FF. The natural history and treatment of adrenal myelolipoma. J Urol 1997;157(4):1213-1216.
pubmed doi - Ravichandran R, Lafferty F, McGinniss MJ, Taylor HC. Congenital adrenal hyperplasia presenting as massive adrenal incidentalomas in the sixth decade of life: report of two patients with 21-hydroxylase deficiency. J Clin Endocrinol Metab 1996;81(5):1776-1779.
pubmed doi - Hagiwara H, Usui T, Kimura T, Tagami T, Naruse M, Minamiguchi S, Kato T, et al. Lack of ACTH and androgen receptor expression in a giant adrenal myelolipoma associated with 21-hydroxylase deficiency. Endocr Pathol 2008;19(2):122-127.
pubmed doi
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.