

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website http://www.jofem.org |
Case Report
Volume 2, Number 4-5, October 2012, pages 198-204
A Rare Case of De novo C634G Germline Mutation of the RET Proto-Oncogene in a Patient With MEN 2A Syndrome With Aggressive Pheochromocytoma
Laurentiu M. Popa, d, Shirley X. Yanb, Ildiko Lingvayc
aCancer Immunobiology Center, University of Texas Southwestern Medical Center at Dallas, 6000 Harry Hines Blvd., NB 9.228, Dallas, Texas, USA
bDepartment of Pathology, University of Texas Southwestern Medical Center at Dallas, 5909 Harry Hines Blvd., HA02.171A, Dallas, Texas, USA
cDivision of Endocrinology, University of Texas Southwestern Medical Center at Dallas, 5323 Harry Hines Blvd., U9.134E, Dallas, Texas, USA
dCorresponding author: Laurentiu M. Pop, Cancer Immunobiology Center, University of Texas Southwestern Medical Center at Dallas, 6000 Harry Hines Blvd., Dallas, Texas, USA
Manuscript accepted for publication October 1, 2012
Short title: RET Proto-Oncogene
doi: https://doi.org/10.4021/jem120w
| Abstract | ▴Top |
Multiple endocrine neoplasia type 2A (MEN 2A) is an autosomal inherited disorder induced by mutations in theRET proto-oncogene. It is characterized by medullary thyroid carcinoma (MTC), accompanied in half of cases by pheochromocytoma and less frequently, by primary hyperparathyroidism. The aim of the study was to investigate a clinical case of a MEN 2A syndrome consisting of bilateral aggressive pheochromocytomas and MTC with loco-regional lymph node invasion caused by a rare de novo germline mutation in the RET proto-oncogene (Cys634Gly). We reported herein a 42-year-old man presented with hypertension refractory to various antihypertensive drug regimens administered over the preceding 3 years. Abdominal computed tomography scan showed a large right adrenal mass, and a smaller left adrenal nodule. Comprehensive biochemical analyses (24-hour urinary catecholamines and metabolites) confirmed the diagnosis of pheochromocytoma. 123I-metaiodobenzylguanidine scintigraphy revealed uptake in the left inferior thyroid nodule. Left thyroid ultrasound-guided fine needle aspiration established the diagnosis of MTC. The patient first underwent open right adrenalectomy, and 2 months later, total thyroidectomy with central neck compartment lymph node dissection.Screening for hyperparathyroidism was negative. On follow-up, the patient demonstrated no biochemical abnormalities, or clinical complaints. Genetic testing identified a point mutation in the RET proto-oncogene, confirming the diagnosis of MEN 2A. This missense mutation, located in exon 11, resulted in the replacement of a cysteine 634 residue with glycine.
To the best of our knowledge, this is the first MEN 2A case reported with de novo Cys634Gly RET germline mutation in North America, and also worldwide with regard to the aggressiveness of this genotypic variant.
Keywords: MEN 2A; RET proto-oncogene Cys634Gly mutation; Aggressive pheochromocytoma
| Introduction | ▴Top |
Multiple endocrine neoplasia type 2 (MEN 2) is an autosomal inherited syndrome caused by germline activating mutations in the Rearranged during transfection (RET) proto-oncogene. Currently, three distinct subtypes have been described based on clinical appearance: MEN 2A, MEN 2B, and familial medullary thyroid carcinoma (FMTC). The common endocrinopathy is medullary thyroid carcinoma (MTC), as all the patients affected by the MEN 2 genetic condition develop thyroid malignancy in their lifetime (100% penetrance).
MEN 2A, known also as Sipple syndrome, implies the occurrence of a pheochromocytoma (20-60% penetrance; either benign or malignant) and a primarily benign hyperparathyroidism (HPT) in 10-30% of cases, with very rare malignancy reported [1, 2]. Patients with MEN 2B syndrome do not develop parathyroid disease, but a particular phenotype including a marfanoid habitus, multiple mucosal ganglioneuromas, neurofibromas, and thickened corneal nerves define this syndrome. FMTC is the least aggressive subtype and is characterized only by MTC, inherited in an autosomal dominant fashion with a genetic penetrance of nearly 100% [3]. MEN 2 has a prevalence of nearly 2.5 per 100,000 in the general population, with the distribution of the three subtypes being 70-80% MEN 2A, 5% MEN 2B and 10-20% FMTC [4]. Genetic tests for mutations have revealed a complex polymorphism in codons for both extra- and intracellular domains of the protein product of the RET gene, the only gene demonstrated to be responsible for MEN 2 development. The most frequently affected residue is cysteine 634, located in exon 11, which has been found to be mutated in 87% of patients with MEN 2A syndrome [5].
We report here the diagnosis of MEN 2A syndrome in a patient in his fifth decade of life, manifesting as bilateral aggressive phechromocytomas and MTC with loco-regional lymph node invasion and caused by an uncommon de novo germline mutation in the RET proto-oncogene (Cys634Gly).
| Case Report | ▴Top |
A 42-year-old man with normal body mass index was presented in our endocrinology clinic with a history of high blood pressure refractory to several antihypertensive regimens for 3 years. He described episodes characterized by tremors, intermittent occipital headaches and palpitations. Random outpatient measurements of blood pressure showed several readings with systolic blood pressure above 200 mmHg with low-normal pulse rate. The frequency of these episodes had gradually increased over the preceding three years, becoming daily prior to presentation. He had no other complaints and felt normal between episodes.
There was no family history of thyroid or parathyroid problems, nor pheochromocytoma. His father is alive and well at age 68, and has well-controlled hypertension. His mother is alive and well at age 65, with no known medical problems. His paternal grandfather died at age 75 of heart disease. He has one 40 year-old sister, who is alive and well. He has two sons, ages 11 and 16, with no medical problems. His past medical history consisted only of hyperlipidemia, treated with a statin.
The refractory hypertension, along with the episodes described above, prompted an abdominal computed tomography (CT) scan, which revealed the presence of a large right adrenal mass measuring 8.3 × 7.4 × 7.1 cm, and a 1.5 cm left adrenal nodule. Subsequent biochemical evaluation of adrenal function showed elevated 24-hour urinary catecholamines and metabolites (norepinephrine 10-fold above normal range, epinephrine 20-fold, normethanephrine and methanephrine 20-fold, and vanillylmandelic acid (VMA) 6-fold) and were consistent with a diagnosis of pheochromocytoma (Table 1). Plasma renin, serum aldosterone, dehydroepiandrosterone, serum and urinary corisol, urinary dopamine, free T3, free T4 and TSH were normal.
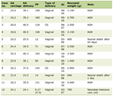 Click to view | Table 1. Dynamic Levels of 24-Hours Urinary Excretion of Catecholamines and Their Metabolites |
To localize the tumor, 123I-metaiodobenzylguanidine scintigraphy (MIBG) was performed and revealed uptake in both adrenal glands, on the right side corresponding with the 8.3 cm adrenal mass, and on the left side corresponding with the 1.5 cm adrenal nodule seen on the CT scan. A left inferior thyroid nodule with MIBG uptake was also observed. Thyroid ultrasound-guided fine needle aspiration of the left thyroid showed smears composed of cellular fragments and clusters of atypical follicular cells. The cells exhibited enlarged, oval-shaped nuclei, prominent nucleoli, intranuclear inclusion and nuclear pallor. The patient’s calcitonin level was 1,174 pg/mL (reference range < 10 pg/mL). A diagnosis of MTC was made based on these findings, which in the context of a diagnosed pheochromocytoma, indicated the clinical diagnosis of MEN 2A. Subsequent genetic testing identified the following point mutation in the RET proto-oncogene: p.C634G (c.1900T > G), a missense mutation in a cysteine codon encoded in exon 11 (codon 634, TGC → GGC, Cys → Gly).
A decision was made to proceed with removal of the larger adrenal lesion only (right adrenalectomy), in order to minimize surgical risk and also to postpone for as long as clinically feasible the ensuing adrenal insufficiency. After adequate alpha-blockade with phenoxybenzamine (followed by beta-blockade with propranolol) and preoperative salt and fluid loading to prevent intraoperative blood pressure changes, the patient underwent open right adrenalectomy. Gross examination of the right adrenal gland showed three well circumscribed masses grossly arising from the medulla with the largest mass measuring 8.0 × 8.0 × 6.0 cm (Fig. 1A). Two other masses measure 3.6 × 2.3 × 1.5 cm and 2.0 × 1.5 × 1.2 cm. The microscopic examination revealed pheochromocytomas with predominantly alveolar (zellballen) and trabecular growth pattern. The pheochromocytomas have focally diffused growth areas and large nests (Fig. 1A). There are areas of spindle-shaped tumor cells (Fig. 1B, C) and marked nuclear pleomorphism (Fig. 1D). A monotonous tumor cell population was noted in focal areas, with smaller tumor cells and a high nuclear to cytoplasmic ratio observed. Mitotic figures were increased up to 4 per 10 high power fields (magnification at × 40 with a × 10 objective lens using an Nikon Eclipse 80i microscope) (Fig. 1C). No definitive evidence of capsular invasion was observed but the presence of foci of small tumor nests covered by endothelium in the large capsular/septal vessels strongly suggested vascular invasion. Taken together, the Pheochromocytoma of the Adrenal gland Scaled Score (PASS) [6] was considered to be ≥ 7. The Ki-67 proliferative index was 5% (Fig. 2) and the S100 protein was absent in diffuse growth areas.
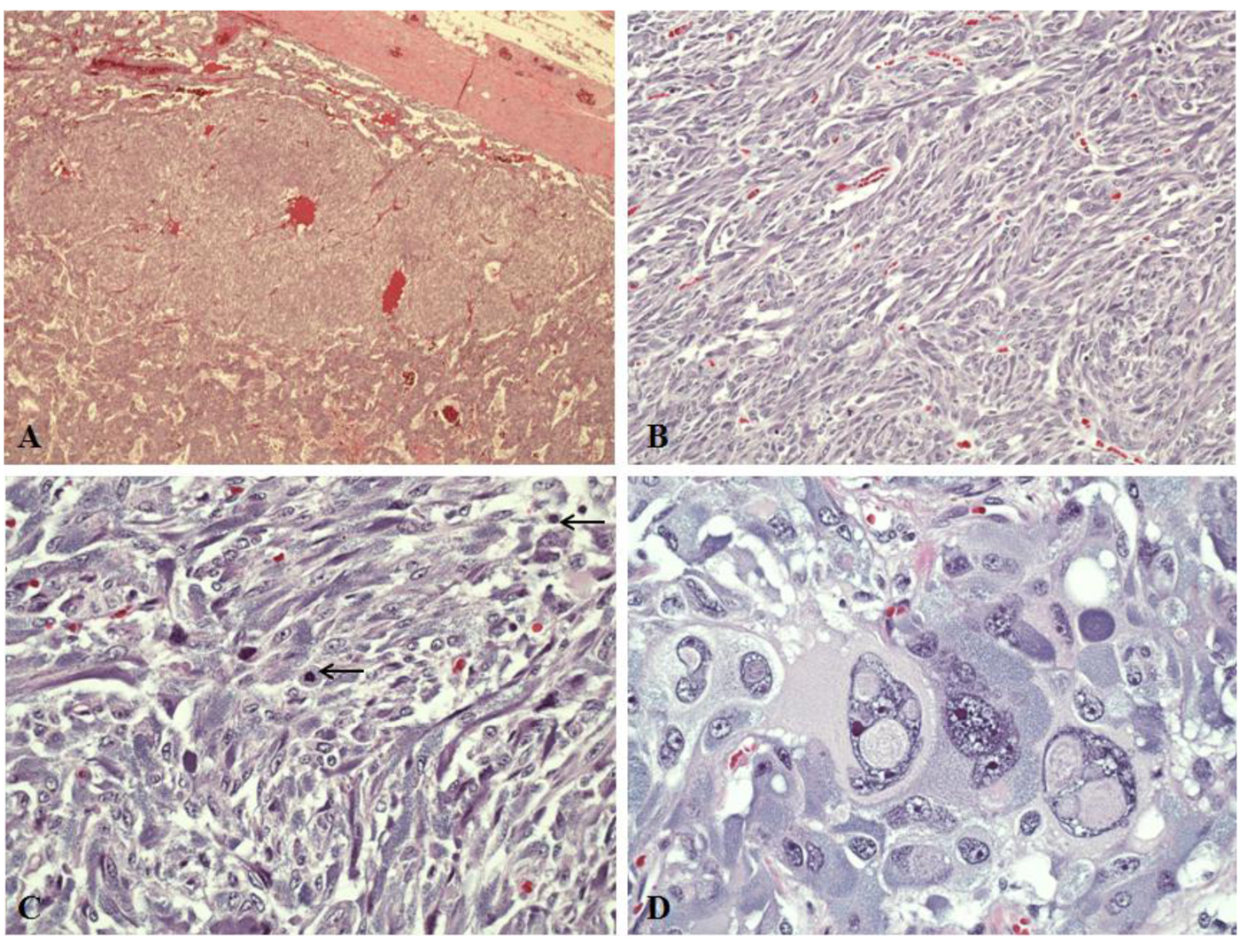 Click for large image | Figure 1. Light microscopy in pheochromocytomas of right adrenal gland (hematoxylin and eosin stains). A: A large tumor nest surrounded by tumor in the “zellballen” growth pattern (magnification, × 4). B: Diffuse growth area showing spindle-shaped tumor cells (magnification, × 20). C: Mitotic figures (black arrows) are observed in the spindly growth area (magnification, × 40). D: Tumor cells with profound nuclear pleomorphism (magnification, × 40). |
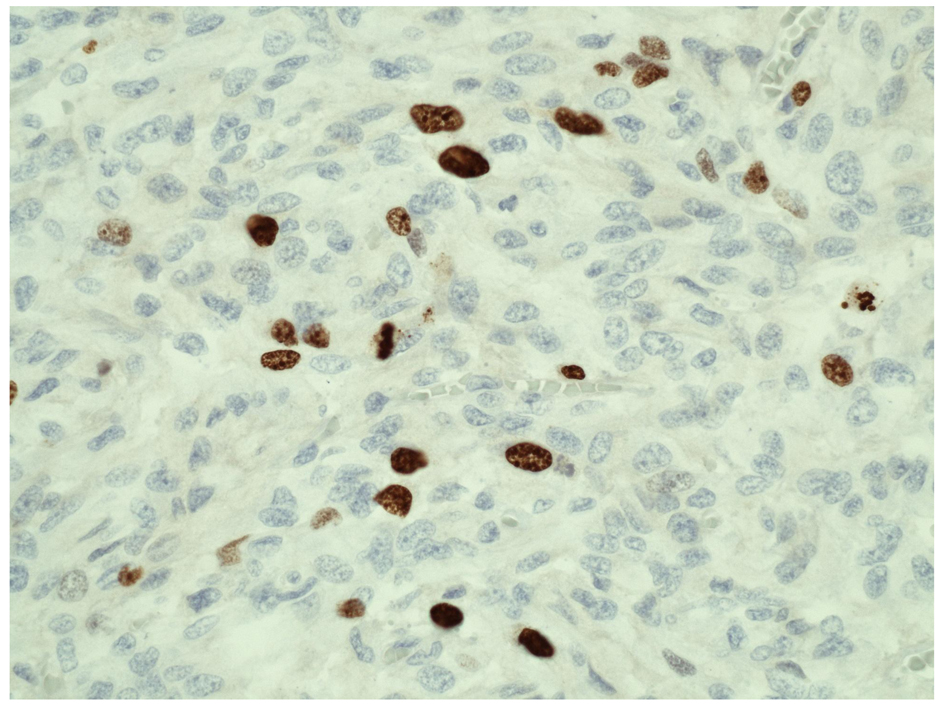 Click for large image | Figure 2. Ki-67 immunohistochemical study of pheochromocytoma (right adrenal gland). Ki-67 (MIB-1) proliferation index shows increased proliferative activity (> 5%) (magnification, × 40). |
At 3 weeks postoperatively, the patient’s blood pressure was 149/80 off all hypertensive medications, and urinary metanephrines and VMA were 2-fold higher than the normal range, but with normal levels of plasma fractionated free metanephrines (Table 1, 2). Thus, alpha-adrenergic blockade was indicated before thyroid surgery, which was performed two months post-adrenalectomy. The patient underwent total thyroidectomy, bilateral central (level VI) lymph node dissection, and excisional biopsy of two pre-operatively identified right lower neck nodules. Macroscopically, the right lobe was 4.6 × 2.5 × 1.0 cm with a single firm, irregular, yellow-tan mass localized in its inferior pole measuring 1.0 × 0.8 × 0.6 cm. The left lobe was 5.0 × 2.5 × 0.7 cm, and sectioning revealed a 2.0 × 1.4 × 1.3 cm yellow-tan mass comprising the majority of the corresponding inferior pole. Microscopy confirmed a well-defined lesion characterized by proliferation of nests and cords of neoplastic spindle to epitheloid cells situated in areas of a hyalinized stroma. The immunostaining for calcitonin was positive. The histopathological features and immunostaining pattern confirmed multifocal, moderately differentiated MTC involving the left lobe and inferior aspect of the right lobe. Sections made from 2 out of 5 lymph nodes localized in the central compartment demonstrated nests of atyipical cells within a desmoplastic stroma that also stained positive for calcitonin. Both right lower neck nodules were calcitonin-negative, without any pathological sign of MTC invasion. The pathologic extent of disease (pTNM) was noted as pT1b (tumor 1 - 2 cm, limited to thyroid), pN1A (2 out of 5 level VI lymph nodes without extracapsular extension). Thyroid hormone replacement was started the next day after surgery and thyroid function tests remained normal at subsequent follow-ups. Three and six months postoperatively, the calcitonin level was 17 pg/mL (both times), and a neck sonogram performed six months postoperatively showed no evidence of loco-regional recurrence of the MTC.
Click to view | Table 2. Postoperative Levels of Plasma Fractionated Free Metanephrines |
Screening for hyperparathyroidism was carried out several times, prior to surgery as well as during subsequent follow-ups, and was negative. At six months after right adrenalectomy, the abdominal CT scan demonstrated no signs of right local recurrence. The left adrenal gland showed nodular thickening of the limbs which appeared more rounded than in the initial CT examination, but no significant enhancement was noted. The levels of 24-Hours urinary excretion of catecholamines and their metabolites, and plasma fractionated free metanephrines were within the reference ranges (Table 1, 2). Radiological follow-up along with biochemical evaluation will be performed every 6 months to assess both the progression of the left adrenal lesion, as well as surveillance for regional or distant recurrence of this aggressive pheochromocytoma.
Genetic screening of the patient’s family identified the mutation in one of his two sons, who is 11 years old (older than 5 years, the recommended age for prophylactic thyroidectomy). He had negative screening for MTC, HPT, and pheocromocytoma and underwent prophylactic total thyroidectomy. The patient’s mother tested negative and testing for the father was deemed unnecessary by his primary physician, based on the results of biochemical testing.
| Discussion | ▴Top |
The RET gene is localized on chromosome 10 and encodes a tyrosine kinase receptor with the critical function of neural crest development and differentiation [7]. Its ligands are members of the glial cell line-derived neurotrophic factor family, which act mainly as growth factors. Loss of function mutations in the RET gene cause Hirschsprung’s disease, an abnormal development of the enteric nervous system, which leads to colon aganglionosis. When gain of function mutations occur in the RET gene, congenital and de novo malignancies of the neuroendocrine system develop (MTC, MEN 2 disorders). The association of germline mutations of the RETproto-oncogene with the MEN 2 phenotype represents the first proven genotype-phenotype correlation within the etiology of any inherited malignant disease [8].
The majority of hotspot codons affected within gain of function mutations in MEN 2A were found in the cysteine residues in exons 10 (codons 609, 611, 618 and 620) and 11 (codons 630 and 634) encoding the extracellular domain of the RET molecule [8, 9]. Interestingly, the genotype-phenotype correlation of RET mutations with the MEN 2A syndrome was most significant for mutations of the cysteine codon 634 which was also correlated with the highest prevalence of pheochomocytoma and HPT [8, 10]. To date there are 6 types of mutations described in the cysteine 634 residue in the following order of frequency: Arg, Tyr, Gly, Phe, Trp and Ser. The most penetrant mutation, which also confers the most aggressive phenotype, was found to be Cys634 Arg [8, 10, 11]. This feature was further supported by another study, which showed that the patients with Cys634Arg mutation had more distant MTC metastases at the time of diagnosis than those with the Cys634Tyr or Cys634Trp mutations and that the metastases developed earlier in patients with Cys634Arg mutations than in those affected by Cys634Tyr mutation [12]. The very low number of identified cases of families with the Cys634Gly mutation makes it improbable to predict the phenotypic and clinical behavior determined by this mutation, despite the fact that one study has suggested that this unique mutation might be associated with early MTC development and high consecutive risk of lymph nodes dissemination [13]. In spite of fact that the mutations in codon 634 are, by far, the most frequent reported, there are only a few cases with C634G mutations described in Germany [13, 14], the Netherlands [13, 15], Italy [16, 17], Romania [18], India [19] and Iran [20, 21], but mainly among patients with hereditary MTC or MEN2A without pheochromocytoma. Interestingly, a very high incidence of C634G germline mutation (5 cases out of 43) was observed in Iranian patients with hereditary MTC, indicating that the observed C634G might represent a founder effect [22]. However, another study also of Iranian patients with hereditary MTC published in the same year did not report any C634G mutations, which may be related to the different genetic background of the patients involved in the study [21].
In the United States, a retrospective study performed by Quayle et al on a cohort of 323 patients with MEN 2A syndrome and known RET mutation showed that < 1% (3 out of 323 patients) harbored a C634G mutation, with no accompanying pheochromocytoma [23]. Although the authors suggested an association between pheochromocytoma occurrence and codon 618 mutations, they concluded that there are many limitations to their study (number of kindreds or presence of less frequent mutations). However, their study strongly supported that the screening for pheochromocytoma in MEN 2A family members should be done starting at 10 years of age. MEN2A cases with C634G mutation and consisting of MTC with pheochromocytoma are very rarely presented in literature, with one case in France [24], one in Italy [16], and two in Slovenia [25].
Our patient presented with a level 2 RET mutation, which is considered to confer an intermediate risk for earlier development of MTC (along with codons 611, 618, and 620) [26]. Lack of family history of any clinical endocrinopathy belonging to MEN 2A and the confirmed absence of mutation in one parent suggest that the patient had a de novo mutation.
The delineation between benign and malignant features of pheochromocytoma has yet to be determined for our patient. To the best of our knowledge, no Cys634Gly mutation was described for malignant pheochromocytomas in the context of a MEN 2A syndrome. A PASS score > 4 [27] and the size of the largest tumor being > 6 [27] strongly suggest malignant biological behavior. The current guideline is to consider a malignant pheochromocytoma only when metastases are observed [6]. However, recent studies have underlined the necessity of adding new parameters, such as increased Ki-67 index (> 4%) and the absence of S100 protein, for a clearer definition of malignancy [27]. A malignancy diagnosis based on surgical pathology findings from the primary tumor, prior to the occurrence of clinically detectable distant metastasis, could have a significant impact on patient counseling and potentially on treatment selection. Further management of our patient’s pheochromocytoma will be dependent on the progression of his left adrenal gland tumor and/or occurrence of any local or distant metastases. If removal of the left pheochromocytoma is warranted, an open procedure to preserve the cortex of the adrenal would be desirable to avoid adrenal insufficiency.
Although the Cys634 mutations predispose to parathyroid disease, our patient’s calcium and iPTH level have been within normal range. The patient will continue to be screened periodically for HPT, and in the presence of any sign of HPT, total parathyroidectomy with forearm autograft will be indicated.
Earlier it was believed that a Cys634Gly mutation could be associated with lichen planus amyloidosis, a cutaneuos disorder characterized by amyloid deposits in the skin that presents clinically with rashes and pruritus [28]. However, there are studies showing the same dermatologic manifestation in patients from a family sharing the Cys634Arg mutation [29]. Our patient did not show any sign of cutaneous disease.
The recent American Thyroid Association (ATA) guidelines stratified all known RET mutations into one of four risk levels (A-D) [10]. The 634 mutation is considered a high risk ATA type C mutation and the recommendations for the family members who genetically test positive are: prophylactic thyroidectomy before the age 5 years; annual albumin-corrected calcium or ionized serum calcium measurements (with or without serum iPTH) beginning at the age of 8; and annual plasma free metanephrines and normetanephrines or 24-hour urine collection for metanephrines and normetanephrines beginning by age 20. However, any pre- and post-genetic testing must be followed by specialized counseling [11]. Considering that the genetic testing of one of the patient’s sons, who is 11 years old, revealed the inherited C634G mutation, a thyroidectomy was scheduled and follow-up was planned in accordance with above-mentioned ATA guidelines.
The low incidence of de novo mutations still makes it difficult to establish the role of each type of RET mutation (codon and type of aminoacid residues) in the development and progression of MEN 2 syndromes. It is important to mention that due to the low prevalence of MEN 2 syndromes, genetic testing for RET gene mutations in the general population is not currently performed. Moreover, it is possible that the members of a family carrying the same RET mutation display different levels of MEN 2 aggressiveness. This may be due to varying levels of genetic penetrance, the presence of genetic modifiers or even other associated medical conditions that can interfere with the accuracy of genetic testing. Nevertheless, all patients with clinical signs of MTC should be investigated for the hotspot mutations in the RET gene and tested for urinary norepinephrine. However, genetic screening for MEN 2 with only a clinical presentation of pheochromocytoma (no signs of MTC) is highly debatable due to the high number of genes involved in pheochromocytoma etiology (Von-Hippel Lindau, RET, succinate dehydrogenase subunits B, C, D and neurofibromatosis type 1 genes) [30] and the high cost of genetic testing.
This is the first published case report of a patient with a de novo Cys634Gly RET germline mutation in North America. Moreover, the described mutation induced a very aggressive phenotype, which has not been previously documented in any of the worldwide reports of MEN 2A syndrome caused by a C634G RET mutation.
Acknowledgments
We gratefully acknowledge Drs. Anjanette Tan from Diabetes and Thyroid Center Forth Worth, TX, Erin Dunnigan-Roe, and Fiemu Nwariaku, both from UT Southwestern at Dallas, TX, for their excellent medical and surgical care provided to the patient. We thank Dr. Carol Adair from Baylor Medical Center at Dallas, TX, for her critical review of the histopatological specimens of pheochromocytoma.
Disclosure Summary
The authors have nothing to disclose.
| References | ▴Top |
- Alfaro JJ, Lamas C, Estrada J, Lucas T. MEN-2A syndrome and pulmonary metastasis. Postgrad Med J. 2002;78(915):51-52.
doi pubmed - Jenkins PJ, Satta MA, Simmgen M, Drake WM, Williamson C, Lowe DG, Britton K, et al. Metastatic parathyroid carcinoma in the MEN2A syndrome. Clin Endocrinol (Oxf). 1997;47(6):747-751.
doi - Farndon JR, Leight GS, Dilley WG, Baylin SB, Smallridge RC, Harrison TS, Wells SA, Jr. Familial medullary thyroid carcinoma without associated endocrinopathies: a distinct clinical entity. Br J Surg. 1986;73(4):278-281.
doi pubmed - Moline J, Eng C. Multiple endocrine neoplasia type 2: an overview. Genet Med. 2011;13(9):755-764.
doi pubmed - Cuccuru G, Lanzi C, Cassinelli G, Pratesi G, Tortoreto M, Petrangolini G, Seregni E, et al. Cellular effects and antitumor activity of RET inhibitor RPI-1 on MEN2A-associated medullary thyroid carcinoma. J Natl Cancer Inst. 2004;96(13):1006-1014.
doi pubmed - Thompson LD. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002;26(5):551-566.
doi pubmed - Airaksinen MS, Holm L, Hatinen T. Evolution of the GDNF family ligands and receptors. Brain Behav Evol. 2006;68(3):181-190.
doi pubmed - Eng C, Clayton D, Schuffenecker I, Lenoir G, Cote G, Gagel RF, van Amstel HK, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA. 1996;276(19):1575-1579.
doi pubmed - Ponder BA. Mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2. Cancer Surv. 1995;25:195-205.
pubmed - Yip L, Cote GJ, Shapiro SE, Ayers GD, Herzog CE, Sellin RV, Sherman SI, et al. Multiple endocrine neoplasia type 2: evaluation of the genotype-phenotype relationship. Arch Surg. 2003;138(4):409-416; discussion 416.
doi pubmed - Kloos RT, Eng C, Evans DB, Francis GL, Gagel RF, Gharib H, Moley JF, et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid. 2009;19(6):565-612.
doi pubmed - Punales MK, Graf H, Gross JL, Maia AL. RET codon 634 mutations in multiple endocrine neoplasia type 2: variable clinical features and clinical outcome. J Clin Endocrinol Metab. 2003;88(6):2644-2649.
doi pubmed - Machens A, Niccoli-Sire P, Hoegel J, Frank-Raue K, van Vroonhoven TJ, Roeher HD, Wahl RA, et al. Early malignant progression of hereditary medullary thyroid cancer. N Engl J Med. 2003;349(16):1517-1525.
doi pubmed - Machens A, Dralle H. Familial prevalence and age of RET germline mutations: implications for screening. Clin Endocrinol (Oxf). 2008;69(1):81-87.
doi pubmed - Landsvater RM, Jansen RP, Hofstra RM, Buys CH, Lips CJ, Ploos van Amstel HK. Mutation analysis of the RET proto-oncogene in Dutch families with MEN 2A, MEN 2B and FMTC: two novel mutations and one de novo mutation for MEN 2A. Hum Genet. 1996;97(1):11-14.
doi pubmed - Elisei R, Romei C, Cosci B, Agate L, Bottici V, Molinaro E, Sculli M, et al. RET genetic screening in patients with medullary thyroid cancer and their relatives: experience with 807 individuals at one center. J Clin Endocrinol Metab. 2007;92(12):4725-4729.
doi pubmed - Romei C, Mariotti S, Fugazzola L, Taccaliti A, Pacini F, Opocher G, Mian C, et al. Multiple endocrine neoplasia type 2 syndromes (MEN 2): results from the ItaMEN network analysis on the prevalence of different genotypes and phenotypes. Eur J Endocrinol. 2010;163(2):301-308.
doi pubmed - Paun DL, Mohora M, Duta C, Dumitrache C. Genetic testing for multiple endocrine neoplasia type 2. Rom J Intern Med. 2008;46(2):159-163.
pubmed - Sharma BP, Saranath D. RET gene mutations and polymorphisms in medullary thyroid carcinomas in Indian patients. J Biosci. 2011;36(4):603-611.
doi - Hedayati M, Nabipour I, Rezaei-Ghaleh N, Azizi F. Germline RET mutations in exons 10 and 11: an Iranian survey of 57 medullary thyroid carcinoma cases. Med J Malaysia. 2006;61(5):564-569.
pubmed - Alvandi E, Akrami SM, Chiani M, Hedayati M, Nayer BN, Tehrani MR, Nakhjavani M, et al. Molecular analysis of the RET proto-oncogene key exons in patients with medullary thyroid carcinoma: a comprehensive study of the Iranian population. Thyroid. 2011;21(4):373-382.
doi pubmed - Hedayati M, Zarif Yeganeh M, Sheikhol Eslami S, Rezghi Barez S, Hoghooghi Rad L, Azizi F. Predominant RET Germline Mutations in Exons 10, 11, and 16 in Iranian Patients with Hereditary Medullary Thyroid Carcinoma. J Thyroid Res. 2011;2011:264248.
- Quayle FJ, Fialkowski EA, Benveniste R, Moley JF. Pheochromocytoma penetrance varies by RET mutation in MEN 2A. Surgery. 2007;142(6):800-805; discussion 805 e801.
- Sanz C, Vezzosi D, Pigny P, Bennet A, Caron P. Multiple endocrine neoplasia type 2a and germ line C634G RET mutation diagnosed in an 80-year-old patient. Ann Endocrinol (Paris). 2009;70(2):141-144.
doi pubmed - Bergant D, Hocevar M, Besic N, Glavac D, Korosec B, Caserman S. Hereditary medullary thyroid cancer in Slovenia—genotype-phenotype correlations. Wien Klin Wochenschr. 2006;118(13-14):411-416.
doi pubmed - Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, Conte-Devolx B, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. 2001;86(12):5658-5671.
doi pubmed - de Wailly P, Oragano L, Rade F, Beaulieu A, Arnault V, Levillain P, Kraimps JL. Malignant pheochromocytoma: new malignancy criteria. Langenbecks Arch Surg. 2012;397(2):239-246.
doi pubmed - Seri M, Celli I, Betsos N, Claudiani F, Camera G, Romeo G. A Cys634Gly substitution of the RET proto-oncogene in a family with recurrence of multiple endocrine neoplasia type 2A and cutaneous lichen amyloidosis. Clin Genet. 1997;51(2):86-90.
doi pubmed - Ceccherini I, Romei C, Barone V, Pacini F, Martino E, Loviselli A, Pinchera A, et al. Identification of the Cys634—>Tyr mutation of the RET proto-oncogene in a pedigree with multiple endocrine neoplasia type 2A and localized cutaneous lichen amyloidosis. J Endocrinol Invest. 1994;17(3):201-204.
pubmed - Pawlu C, Bausch B, Reisch N, Neumann HP. Genetic testing for pheochromocytoma-associated syndromes. Ann Endocrinol (Paris). 2005;66(3):178-185.
doi
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.