

| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website http://www.jofem.org |
Review
Volume 2, Number 4-5, October 2012, pages 155-165
Metabolic Syndrome: A Multifaceted Disease of Affluence
Marià Alemanya, b, c
aDepartment of Nutrition and Food Science, Faculty of Biology, University of Barcelona, Barcelona, Spain
bCIBER Nutrition and Obesity, Institute of Health Carlos III, Spain
cCorresponding address: Department of Nutrition and Food Science; Faculty of Biology; University of Barcelona; Av. Diagonal, 645; 08028 Barcelona, Spain
Manuscript accepted for publication October 1, 2012
Short title: Metabolic Syndrome
doi: https://doi.org/10.4021/jem116w
- Abstract
- Introduction
- The Metabolic Syndrome as a Disease
- The Pathognomic Links: Insulin Resistance
- Liver Steatosis
- Obesity
- Hypercortisolism
- Sleep Apnea and Heart Disease
- Nitric Oxide, Hypertension and Blood Flow
- Microbiota Alterations
- Sex Hormones
- Conclusions
- References
| Abstract | ▴Top |
Metabolic syndrome developed in consequence of an evolutionary inadequacy: the human body was unprepared for a dietary excess of nutrients, especially lipids (largely in detriment of carbohydrate). This excess awakens metabolic signals akin to those of starvation, in which the main energy staple is the body’s own lipid reserve. Lipid dietary abundance prevents the use of glucose, which in turn limits the oxidation of amino acids. To ward against a subsequent avalanche of substrates, the immune system and hypertrophied tissues (for example,adipose) elicit a series of defence responses. This response is probably the ultimate basis of a disease that is manifested as various pathologies, which were initially defined as distinct entities but which are slowly being seen as a single pathognomic unit in the literature. Based on their common origin of the ample availability of food in our modern society, the cluster of diseases comprising the metabolic syndrome is probably best described as a single multifaceted disease.
Keywords: Metabolic syndrome; Obesity; Hyperlipidic diet; Inflammation
| Introduction | ▴Top |
The proportion of overweight and obese people is rising unchecked, to epidemic proportions, worldwide. Diabetes, hypertension, and cardiovascular diseases account for most of the world’s metabolism-related disorders, as well as a large share of its total morbidity and mortality. Only cancer, infectious diseases, and violence/accident-related deaths combined surpass these metabolic diseases in terms of mortality, but not in terms of the extent (years) of morbidity or the social and economic aspects affecting the quality of life. Because the association between these and other diseases is not casual, many contemporary authors refer to them conjointly as the metabolic syndrome (MS).
The considerable increase in morbidity and exponential rise in health care and other social costs caused by MS is aggravated by its wide extension across ethnic, economic, and social divides, as well as by its non-responsiveness to attempted remedial measures. The increasing transcendence of MS as one of the main health problems of modern medicine has led to the generation of an enormous mass of related research. Although much of this literature includes reviews and commentary, nevertheless, the full extent of the related research information on MS cannot be fully synthesized / processed by any single investigator.
Is the metabolic syndrome a true syndrome or a multiform disease?
As the number of diseases that may fall under the umbrella term of MS grows, researchers are finding that the pathogenic mechanisms involved in MS are much more complex than previously assumed. As a result, many related research issues have been only sparsely analyzed, whereas others have perhaps been overstudied. This imbalance is partly due to the anisotropic availability of adequate methodology, the medical and social (or commercial) interest of the public, and the availability and rigid compartmentalization of medical specialists.
The purpose of this review is to emphasize the need to include in the working concept of MS a number of associated diseases that are often considered to be related but not crucial to the core of the MS. In other words, the goal is to present MS as a multifaceted disease, rather than as a simple cluster of diseases that are only partially associated but happen to be often, coincidentally, present in patients. This view of MS is not universally shared, probably because the separate diseases (hypertension, obesity, diabetes, etc.) existed prior to the development of the concept of MS. As a result, a corpus of knowledge was developed independently for each disease, and the concept of MS was only defined after researchers sought to find a common relationship or shared origin for the various diseases.
Although individual pathologies obviously correlate with alterations at the molecular level, the aggregation of molecular-level changes and their eventual feedback effects depend on the overall physiological context, for example, the organism as a whole. Only by considering the molecular- and organism-level aspects together can we begin to understand the disease. In other words, both aspects must be considered to discern whether MS is comprised of numerous interconnected, albeit separate, pathologies or whether it is a single hydra-like disease with separate symptoms sharing a common pathogenic core.
Several Health-related Societies and Medical Associations have established detailed protocols for the diagnostic and characterization of the MS, with discrimination limits and algorithms derived from observational studies. However, the pathogenic relationships of the key diseases packed in the broad definition of the MS have not been yet fully established as a single pathognomic unit other than by epidemiological association or the intuitive association of the diseases carried out by a number of experienced and insightful scientists. The MS is a widely recognized cluster of related pathological traits that affect a large proportion of adults in developed and developing societies [1, 2]. Most definitions include insulin resistance/diabetes, hyperlipidemia, arterial hypertension and obesity, often associated with hyperuricemia, small dense LDL, and endothelial inflammation (Table 1).
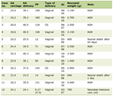 Click to view | Table 1. Main Pathologic Traits Associated With the Metabolic Syndrome |
| The Metabolic Syndrome as a Disease | ▴Top |
Because of the generalized lack of knowledge of the pathogenic origin, and the varying degree of association of its different components, acknowledgement of the MS as a distinct pathologic entity has been slow and progressive. Metabolic syndrome was first discussed in classical times, taking form early last century (reviewed by Sarafidis and Nilsson [3]).
The MS epidemic is generally considered an unwanted consequence of social development and the access to rich, abundant and varied food, decreased physical activity and the other perks (and disadvantages) of modern-day life in most human Societies. The correlations in the incidence of the MS-related diseases have been fully established, as well as their interrelations, mainly from observational analyses; but recent research also uncovered a number of shared mechanisms: genetic, epigenetic, psychosocial, and other mechanisms [4, 5]. Because the pathologic traits share a common (although only partially uncovered) origin [6], researches have indicated that MS should be considered a multifaceted disease. However, the issue is not settled, in part because of the different views on the role of lipid/carbohydrate in the diet as the main factor eliciting MS development [7, 8].
The combination of basic metabolic regulation traits, modulated by specific allelic distributions, epigenetics, and exposures to environmental factors during early development and over one’s lifetime, may cause a wide range of symptom patterns and intensities. Accordingly, MS may be considered a multiform disease. By emphasizing pathogenesis rather than compliance with specific quantitative pathologic indicators, additional alterations are also found to be associated with MS, many of which share common pathways. These alterations can largely be defined as inflammation, and often require a considerable time-span to develop and show up.
It is difficult to determine when MS became a quantitatively significant disease, because we have only had workable definitions for MS within the last few decades. Moreover, most of the components of MS have only recently been able to be fully characterized and diagnosed. Although diabetes, gout, and ictus were described in classical times, obesity was not considered a full disease until recently, although its existence and treatment were described in very old sources [9]. Nevertheless, limited or absent food availability, infection, harsh living conditions, warfare, and exploitation drastically limited human life expectancy during most of the history of mankind, which may have prevented the slowly evolving MS-related pathologies from being manifested in a sufficient number of individuals. Information about people in affluent or privileged positions in various historical contexts showed that they lived longer, but many suffered obesity, gout, and other MS-related diseases, such as arthrosis, hydropesy, ictus, etc. [10]. Therefore, we can assume that MS has been present since historical times, although never before has it affected such huge numbers of individuals. This observation adds weight to its direct relationship with affluence, at least from a nutritional perspective.
| The Pathognomic Links: Insulin Resistance | ▴Top |
A considerable body of literature has established links between the main pathologic traits of MS with the historical precedents indicated above. As the list of related diseases grows, interrelationships continue to be found or proven by direct analysis of the pathogenic pathways or established by association through epidemiological studies.
Many researchers consider insulin resistance to be the main defining element of MS [11]. Insulin resistance is characterized by decreased sensitivity to glucose and low peripheral glucose uptake [12], which often develops into full type-2 diabetes [13]. This metabolic condition has been directly related to neurologic alterations that result in dementia [14], and Alzheimer disease [15]. The metabolic alterations induced by insulin resistance are a direct consequence of the excess availability of dietary lipid. This excess lipid is translated into hyperlipidemia/dyslipoproteinemia [16], with hypertriacylglycerolemia [17], hypercholesterolemia with altered lipoprotein distribution of cholesterol, and small dense LDLs [18]. An obvious consequence of excess lipid energy is a decrement in the overall carbohydrate availability [8] (lipid/carbohydrate energy ratio), which also affects the handling of dietary protein and the excretion of N [19]. This picture is completed by an increased proportion of oxidized lipoproteins in the plasma [20].
Excess lipid-induced insulin resistance also leaves a large amount of glucose (largely dietary) unused, despite the lower overall carbohydrate intake. The disposal of this excess glucose falls largely on the white adipose tissue (WAT), which is already strained by the excess lipid [7], thereby favoring glycolysis and lactate production [21]. The excess lactate is used by the liver for gluconeogenesis [22] or lipid synthesis [23] (Fig. 1).
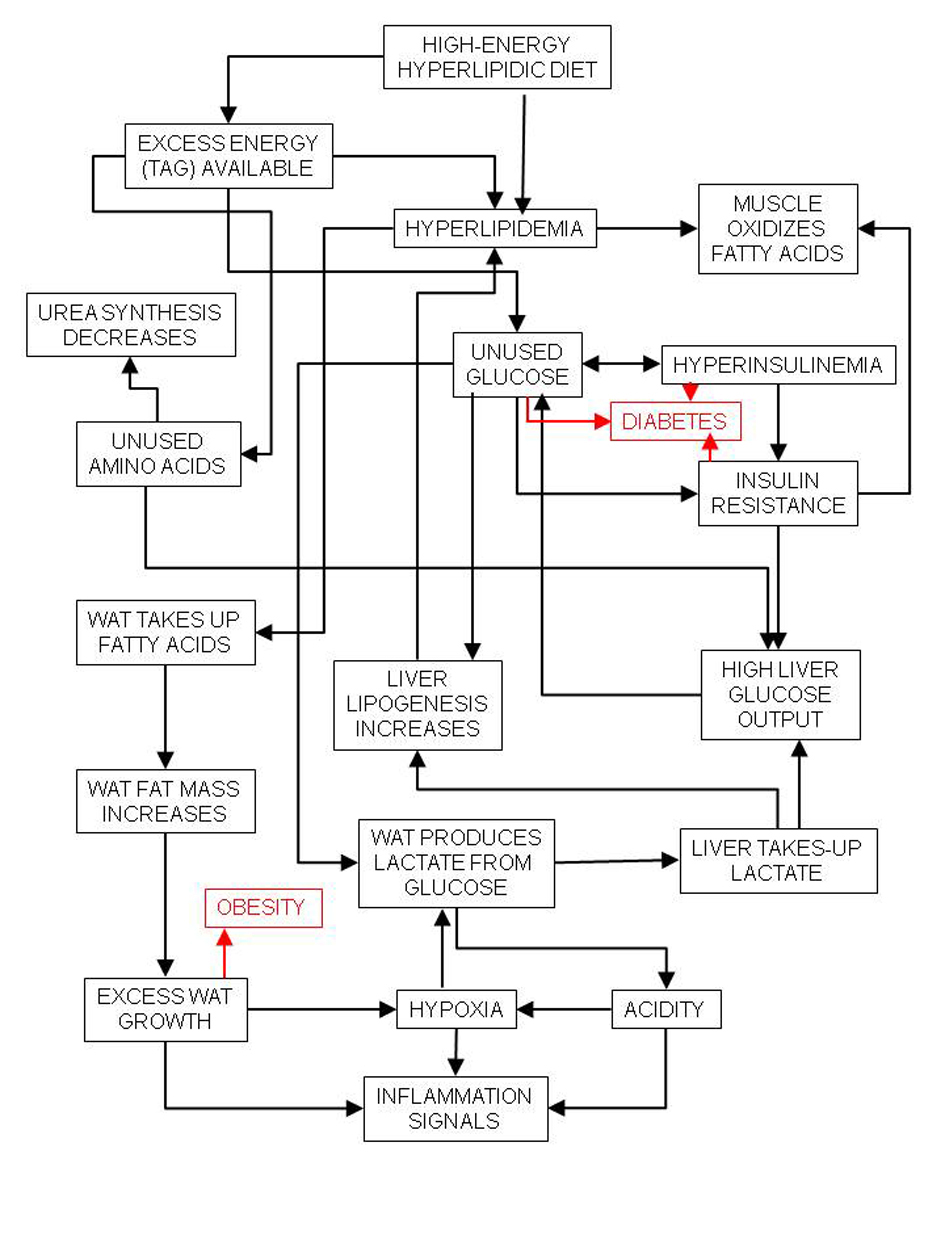 Click for large image | Figure 1. Relationships between high-energy (high-lipid) diet and inflammation and its main consequences, diabetes and obesity. |
Metabolic syndrome also induces changes in the coagulation process of the blood, with altered platelet function[24], and higher reactivity with oxidized lipoproteins [25]. Hypertension enhances the response of the endothelial sheath of blood vessels [26] facilitating the deposit of atheroma plaque [27], favored by hyperlipemia and the inflammatory response of the vessels themselves. A marker of the altered peptide signaling in MS (diabetes) is acanthosis nigricans which is a cutaneous alteration of melanocyte stimulating hormone (MSH) function. In addition to depressive mood, the MS is also related to altered eating behaviors, namely binge-eating [28], and secondarily to reject mechanisms that may in the end result in eating disorders such as anorexia nervosa, bulimia or orthorexia.
| Liver Steatosis | ▴Top |
The excess of lipid availability may overwhelm the liver capacity to process it, eventually resulting in steatosis and hepatomegalia [29]. Consequently, liver functionality decreases and cannot maintain circulating glucose efficiently [30]. The liver is not able to control excess portal insulin [31], and hepatic functions are deeply altered; For example. cells tend to leak enzymes and excess bilirrubin to the plasma [32]. The metabolism of xenobiotics is also affected [33] because of altered defensive ability against oxidative agents [34]. Consequences of these alterations include hyperuricemia [32], gout and arthritis, the latter of which develops because of the enhanced immune response. The circulation of excess lipids favors lipid deposit throughout the tissues, including the muscles and heart [35], as well as lipid utilization from these tissues. Lipid accumulation can diminish the efficiency of the skeletal muscle and heart signal conductivity [36], inducing peripheral nerve damage [37].
| Obesity | ▴Top |
The excess energy availability in the adipocytes, largely driven by insulin resistance elsewhere and excesses of both lipid and glucose (and insulin) in plasma is the key element responsible for white adipose tissue enlargement. This results in obesity, mainly upper body-obesity [38], a consequence of the increase in the size and numbers of adipocytes [39]. The enlargement and continuous availability of energy provoke adipocyte bloating. The cells signal their precarious situation, eliciting an inflammatory response [40], which is largely sustained by the infiltration of immune tissue cells such as macrophages [41]. Excess substrate arrival is in part limited by hypoxic responses [42] driven by glycolysis [43] and decreased blood flow across white adipose tissue, which increases peripheral blood flow resistance and helps induce hypertension [44] (Fig. 2). The altered endocrine/paracrine function of the adipose tissue induces hyperleptinemia and leptin resistance [45], increased circulating levels of resistin and decreasing concentrations of adiponectin and interleukin 6. Increased adipokine signaling also tends to modify overall energy partition and enhance insulin resistance [46].
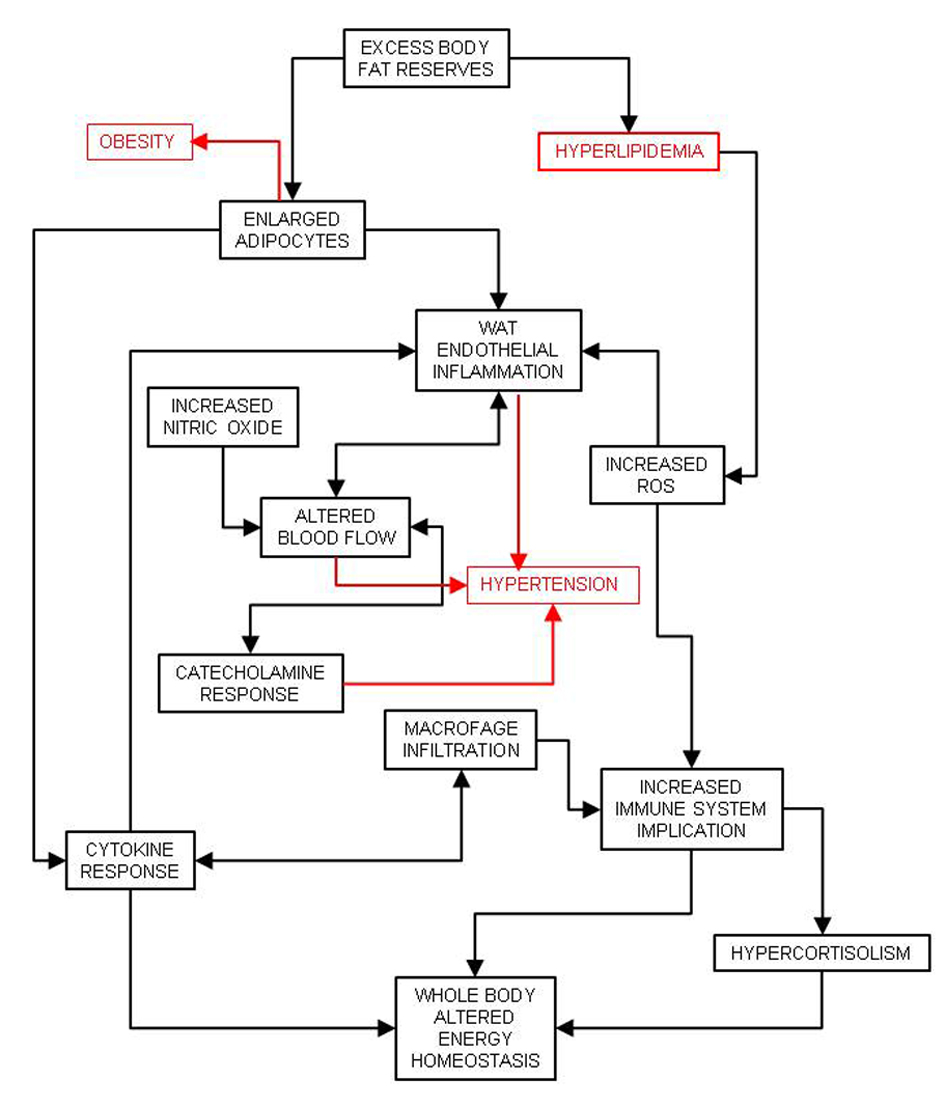 Click for large image | Figure 2. Consequences of white adipose tissue (WAT) defence against excess energy and its relationship with inflammation and hypertension. |
Despite fighting the aggression of the adipocytes by powerful means, the immune system is unable to correct the problem of excess substrate and is unprepared to counteract the tissue damages produced by the excess nutrients. This failure results in the consumption of additional immune resources and the generation of alarm signals. Thus, the inability to eliminate excess nutrients results in collateral damages that can be directly attributed to the increased immune response [47]. These damages can be observed as a higher incidence of asthma [48] or psoriasis [49] in MS patients. The increase in immune response in MS patients may also be related to the development of various autoimmune diseases [50].
| Hypercortisolism | ▴Top |
A consequence of the high immune-system activity is the counter regulatory action of the glucocorticoids. In particular, alterations in the hypothalamic-pituitary-adrenals axis function [51] result in hypercortisolism [52], with conditions close to those seen in Cushing disease. Daily rhythms are flattened or even lost, coincident with metabolic alterations in brain melatonin [53] and, especially, serotonin levels [54]. There is a marked relationship between mood states and the MS, with a high correlation between the syndrome and depressive states [55]. There is also a direct correlation with the duration and quality of sleep [56]. However, it is unclear whether sleep disturbances are a secondary consequence of MS or are, as is depression, a consequence of the alteration of brain function by a cocktail of hormones, cytokines, or available substrates [57]. Metabolic syndrome is also characterized by increased autonomic nervous system activity [57] which affects the response of most tissues to normal nervous regulation.
| Sleep Apnea and Heart Disease | ▴Top |
Sleep apnea is a common symptom of the MS [58]. Although it has been correlated with the main MS pathologies, including obesity, its presentation is largely independent of them [58]. The choking effect of hyperrelaxation of respiratory upper airway muscle has been related to surrounding fat control through paracrine signals. The apnea periods affect the maintenance of a structured sleep [59] and induce spikes of catecholamine secretion that restore breathing, but cause havoc elsewhere. Continued catecholamine bursts may led to atrial fibrillation development, a common finding in the MS [60], and enhance the dangers of hypertension and overall cardiovascular risk. Respiratory insufficiency in obese individuals, compounded by sleep apnea, asthma, and heart malfunction, may result in marked cardiorespiratory dysfunctions that aggravate the metabolic damages in heart muscle contractibility (accumulation of fat, diminished conductivity [35]) and signal strength (atrial fibrillation, pulmonary hypertension [61]). The overall risks of atherosclerosis, hypertension and hypercoagulability, increase considerably the probability of heart failure ictus, and cardiovascular insufficiency [62].
| Nitric Oxide, Hypertension and Blood Flow | ▴Top |
Excess available energy results in diminished protein catabolism, which promotes growth in the young, and helps maintain an active protein turnover [63], as well as a fully functional immune system [64]. However, ammonium production, necessary for urea synthesis and thus N excretion, is limited by this same excess of energy as shown by lower urea excretion [65]. Consequently, patients with MS display excess N availability [19]. Because N does not accumulate in the body, it must be excreted. However, such excretion must occur, at least in part, by mechanisms other than the urea cycle, which is markedly depressed in the MS [65]. Although present at a much reduced scale compared to urea, other excreted forms may include nitric oxide (NO·), its metabolites, nitrate/nitrite [66], but also as nitrogen gas [67].
The increased production of NO·, which is a powerful vasodilator [68], may diminish peripheral resistance to blood flow. However, this effect is readily counteracted by catecholamines and other vasoconstrictors, which aid in hypertension development [69, 70], in response to insulin resistance [71]. Hypertension is also a consequence of increased peripheral resistance due to a higher body mass (in obesity) [72]. However, the main cause of hypertension is the increase in vascular smooth muscle tone [73], due tp signaling through catecholamine, endothelin or angiotensin II [74], which are excreted in part to counteract the increased production of NO·.
The exposure of red blood cells to hypoxia-reoxygenation-derived free radicals [75], NO·, and hyperglycemia (and the consequent increase in protein glycosylation [76]) results in a hardening of the cellular membranes. As a result, in the MS, the red blood cells are less deformable [77] which makes their passage through the capillary beds difficult, increases peripheral resistance to blood flow, and contributes significantly to hypertension [44]. The decreased flexibility of the vessels due to the deposition of atheroma plaques is a critical development of the combination of pathogenic traits described above, developing into full atherosclerosis and hypertension. The changes in blood flow distribution elicited by inflammation result in the extensive distribution of hypoxic areas [78], which may result in mitochondrial damage and endoplasmic reticulum stress [79]. A common parallel finding of these alterations is the overproduction of oxidative radicals, which affect lipoproteins and damage red blood cells and endothelial cells [75], extending and perpetuating the metabolic and functional damages (Fig. 3).
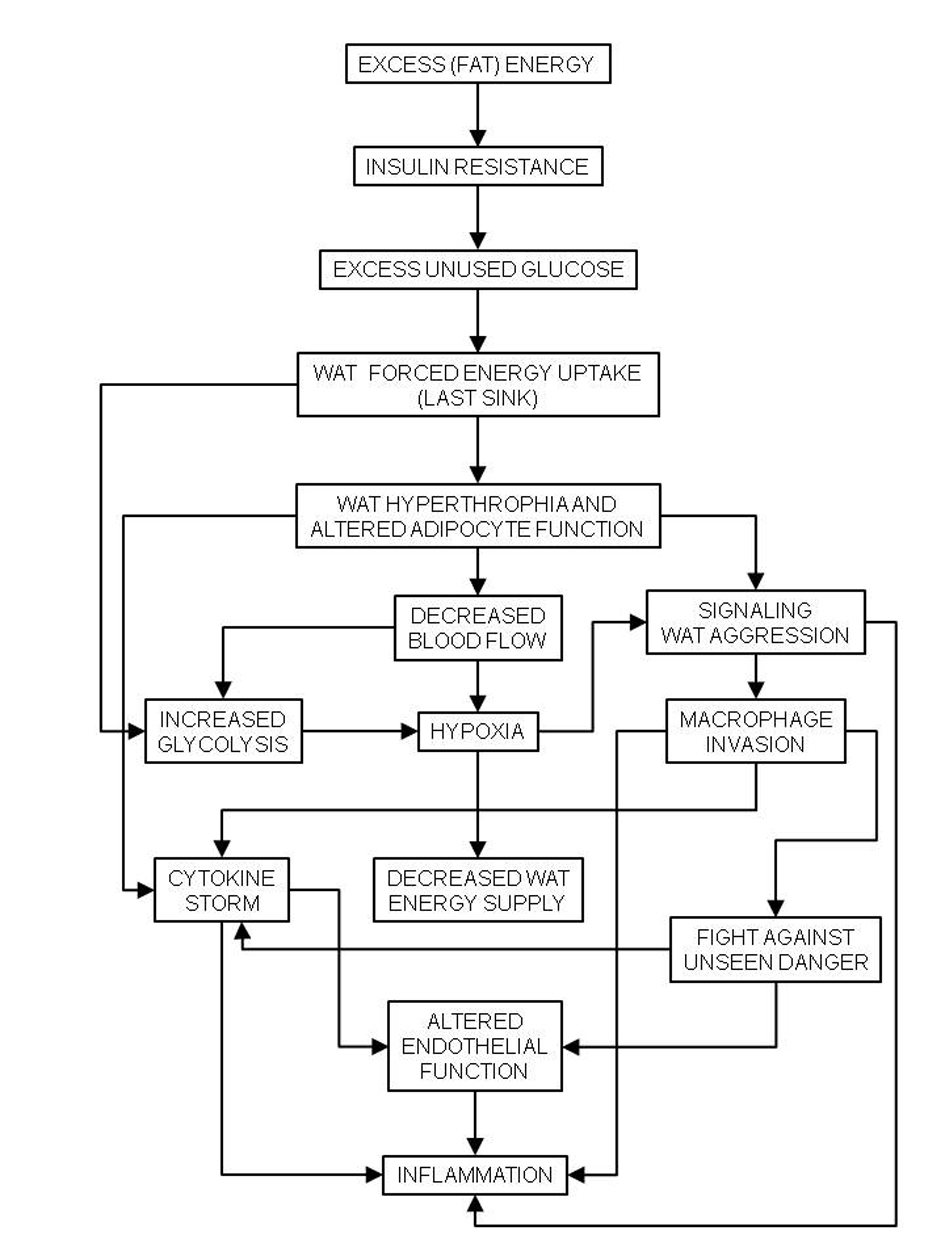 Click for large image | Figure 3. Chain of events caused by hyperlipidic high-energy diets to the last energy sink, white adipose tissue (WAT), causing generalized inflammation, and the basis of the metabolic syndrome. |
| Microbiota Alterations | ▴Top |
Excess NO· synthesis results in the production of large amounts of nitrite and nitrate [66], which are principally excreted though the saliva into the alimentary channel. The presence of nitrates, higher availability of nutrients, and a powerful activated immune system induce functional changes in the composition of the microbiota [80].Such changes may precede the full appearance of MS. The presence of low, albeit maintained, lipopolysaccharide levels in the blood [81] support this implication, and may be sufficient to elicit the described inflammatory response [82]. The reverse may be true as well, but in any case the implication of the microbiota is evident.
| Sex Hormones | ▴Top |
In MS, hypercortisolism develops parallel to a decrease in the levels and activity of androgens [83]. Hypoandrogenism is a consistent finding in male patients with MS [84]. In females, androgen metabolism is also altered, especially in those affected by polycystic ovary syndrome [85]. In all cases, the levels of dehydroepiandrosterone tend to be low [86] because of altered adrenal function, as in advanced age. This finding may be a consequence of alterations in the hypothalamus-pituitary-adrenal (or -gonadal) axes by hyperleptinemia and/or corticosteroids. Decreased estrogen availability specially affects the health of post-menopausal women, since estrogens protect the brain [87] and low estrogen is associated with increased oxidative damage [88]. When compared with glucocorticoids, low sex hormone levels cannot protect sufficiently protein and bone minerals.
The MS has been related to increased incidence of certain cancer types (colon, endometrial, renal cell, gallbladder, upper digestive tract) [89]. In most cases, compared to the effects of MS, these cancers may be more directly related to the diet and, in some cases to the local effects of increased estrogen secretion by abundant adipose and breast tissues [90].
| Conclusions | ▴Top |
The large number of MS-related diseases and pathological traits (Table 1), including some that are derived from the “classical” components of MS, share common origins. All of the aforementioned diseases and traits are inextricably linked, forming a protean “disease of affluence,” the origin of which can be traced to our nutrient-rich living conditions. The diet that our evolution-ingrained desire sought could not be adequately processed by our actual metabolic machinery, which is finely tuned to survive in periods of scarcity but painfully inadequate for extended periods of plenty. Because the cluster of diseases that are directly associated with the MS has a common origin, we believe that it should be considered as a single disease. Like other complex metabolic diseases, the manifestations of MS, which may include the individual pathological traits described above, are consequences of the triple interaction of genetics, environment, and behavior. To this triad we can also include therapeutic (or therapeutic-like) actuations which often backfire into additional iatrogenic damage.
Acknowledgments
This work was supported by grant #SAF2009-11739 from the Plan Nacional de Investigacion en Biomedicina of the Government of Spain.
Competing Interests
The author has no conflicts of interest to disclose.
Abbreviations
MS: metabolic syndrome; WAT: white adipose tissue
| References | ▴Top |
- Sorrentino MJ. Implications of the metabolic syndrome: the new epidemic. Am J Cardiol. 2005;96(4A):3E-7E.
doi pubmed - Desroches S, Lamarche B. The evolving definitions and increasing prevalence of the metabolic syndrome. Appl Physiol Nutr Metab. 2007;32(1):23-32.
doi pubmed - Sarafidis PA, Nilsson PM. The metabolic syndrome: a glance at its history. J Hypertens. 2006;24(4):621-626.
doi pubmed - Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365(9468):1415-1428.
doi - Cornier MA, Dabelea D, Hernandez TL, Lindstrom RC, Steig AJ, Stob NR, Van Pelt RE, et al. The metabolic syndrome. Endocr Rev. 2008;29(7):777-822.
doi pubmed - Bruce KD, Byrne CD. The metabolic syndrome: common origins of a multifactorial disorder. Postgrad Med J. 2009;85(1009):614-621.
doi pubmed - Alemany M. Utilization of dietary glucose in the metabolic syndrome. Nutr Metab (Lond). 2011;8(1):74.
doi pubmed - Volek JS, Feinman RD. Carbohydrate restriction improves the features of Metabolic Syndrome. Metabolic Syndrome may be defined by the response to carbohydrate restriction. Nutr Metab (Lond). 2005;2:31.
doi pubmed - Papavramidou NS, Papavramidis ST, Christopoulou-Aletra H. Galen on obesity: etiology, effects, and treatment. World J Surg. 2004;28(6):631-635.
doi pubmed - Aronson J: Dropsy. Br Med J. 2003;326:491.
doi - Tremblay A. Nutritional determinants of the insulin resistance syndrome. Int J Obes Relat Metab Disord. 1995;19 Suppl 1:S60-68.
pubmed - Rodriguez A, Muller DC, Engelhardt M, Andres R. Contribution of impaired glucose tolerance in subjects with the metabolic syndrome: Baltimore Longitudinal Study of Aging. Metabolism. 2005;54(4):542-547.
doi pubmed - Hanson RL, Imperatore G, Bennett PH, Knowler WC. Components of the "metabolic syndrome" and incidence of type 2 diabetes. Diabetes. 2002;51(10):3120-3127.
doi pubmed - Hashizume K, Suzuki S, Hara M, Komatsu A, Yamashita K. Metabolic syndrome and age-related dementia: endocrinological aspects of adaptation to aging. Mech Ageing Dev. 2006;127(6):507-510.
doi pubmed - Razay G, Vreugdenhil A, Wilcock G. The metabolic syndrome and Alzheimer disease. Arch Neurol. 2007;64(1):93-96.
doi pubmed - Ayyobi AF, Brunzell JD. Lipoprotein distribution in the metabolic syndrome, type 2 diabetes mellitus, and familial combined hyperlipidemia. Am J Cardiol. 2003;92(4A):27J-33J.
doi - Onat A, Sari I, Yazici M, Can G, Hergenc G, Avci GS. Plasma triglycerides, an independent predictor of cardiovascular disease in men: a prospective study based on a population with prevalent metabolic syndrome. Int J Cardiol. 2006;108(1):89-95.
doi pubmed - Satoh N, Wada H, Ono K, Yamakage H, Yamada K, Nakano T, Hattori M, et al. Small dense LDL-cholesterol relative to LDL-cholesterol is a strong independent determinant of hypoadiponectinemia in metabolic syndrome. Circ J. 2008;72(6):932-939.
doi pubmed - Alemany M. The problem of nitrogen disposal in the obese. Nutr Res Rev. 2012;25(1):18-28.
doi pubmed - Holvoet P, Lee DH, Steffes M, Gross M, Jacobs DR, Jr. Association between circulating oxidized low-density lipoprotein and incidence of the metabolic syndrome. JAMA. 2008;299(19):2287-2293.
doi pubmed - DiGirolamo M, Newby FD, Lovejoy J. Lactate production in adipose tissue: a regulated function with extra-adipose implications. FASEB J. 1992;6(7):2405-2412.
pubmed - Mokuda O, Sakamoto Y, Kawagoe R, Shimizu N. Effects of arterial-portal glucose difference on gluconeogenesis from lactate in the isolated bivascular-perfused rat liver. Horm Metab Res. 1993;25(6):285-288.
doi pubmed - Schwarz JM, Linfoot P, Dare D, Aghajanian K. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am J Clin Nutr. 2003;77(1):43-50.
pubmed - Vaduganathan M, Alviar CL, Arikan ME, Tellez A, Guthikonda S, DeLao T, Granada JF, et al. Platelet reactivity and response to aspirin in subjects with the metabolic syndrome. Am Heart J. 2008;156(5):1002 e1001-1002 e1007.
- Meerarani P, Badimon JJ, Zias E, Fuster V, Moreno PR. Metabolic syndrome and diabetic atherothrombosis: implications in vascular complications. Curr Mol Med. 2006;6(5):501-514.
doi pubmed - Rizvi AA. Cytokine biomarkers, endothelial inflammation, and atherosclerosis in the metabolic syndrome: emerging concepts. Am J Med Sci. 2009;338(4):310-318.
doi pubmed - Mc LN. The pathogenesis of atheroma. Am J Pathol. 1952;28(3):413-435.
- Roehrig M, Masheb RM, White MA, Grilo CM. The metabolic syndrome and behavioral correlates in obese patients with binge eating disorder. Obesity (Silver Spring). 2009;17(3):481-486.
doi pubmed - Mendez-Sanchez N, Chávez-Tapia NC, Medina-Santillán R, Villa AR, Sánchez-Lara K, Ponciano-Rodriguez G, Ramos MH, et al. The efficacy of adipokines and indices of metabolic syndrome as predictors of severe obesity-related hepatic steatosis. Dig Dis Sci. 2006;51(10):1716-1722.
doi pubmed - Pagano G, Pacini G, Musso G, Gambino R, Mecca F, Depetris N, Cassader M, et al. Nonalcoholic steatohepatitis, insulin resistance, and metabolic syndrome: further evidence for an etiologic association. Hepatology. 2002;35(2):367-372.
doi pubmed - Wiesenthal SR, Sandhu H, McCall RH, Tchipashvili V, Yoshii H, Polonsky K, Shi ZQ, et al. Free fatty acids impair hepatic insulin extraction in vivo. Diabetes. 1999;48(4):766-774.
doi pubmed - Evangelopoulos AA, Vallianou NG, Panagiotakos DB, Georgiou AT, Zacharias GA, Vogiatzakis ED, Avgerinos PC: The association between uric acid and hepatic function markers with the metabolic syndrome in middle-aged, overweight, and obese people. Endocrinologist 2010;20:312-315.
doi - De Craemer D, Pauwels M, Van den Branden C. Alterations of peroxisomes in steatosis of the human liver: a quantitative study. Hepatology. 1995;22(3):744-752.
doi pubmed - Malinská H, Oliyarnyk O, Hubová M, Zidek V, Landa V, Simáková M, Mlejnek P, et al. Increased liver oxidative stress and altered PUFA metabolism precede development of non-alcoholic steatohepatitis in SREBP-1a transgenic spontaneously hypertensive rats with genetic predisposition to hepatic steatosis. Mol Cell Biochem. 2010;335(1-2):119-125.
doi pubmed - Holloway GP, Snook LA, Harris RJ, Glatz JF, Luiken JJ, Bonen A. In obese Zucker rats, lipids accumulate in the heart despite normal mitochondrial content, morphology and long-chain fatty acid oxidation. J Physiol. 2011;589(Pt 1):169-180.
doi pubmed - Young ME, Guthrie PH, Razeghi P, Leighton B, Abbasi S, Patil S, Youker KA, et al. Impaired long-chain fatty acid oxidation and contractile dysfunction in the obese Zucker rat heart. Diabetes. 2002;51(8):2587-2595.
doi pubmed - Vinik A, Ullal J, Parson HK, Casellini CM. Diabetic neuropathies: clinical manifestations and current treatment options. Nat Clin Pract Endocrinol Metab. 2006;2(5):269-281.
doi pubmed - Bosello O, Zamboni M. Visceral obesity and metabolic syndrome. Obes Rev. 2000;1(1):47-56.
doi - Dulloo AG. Adipose tissue plasticity in catch-up-growth trajectories to metabolic syndrome: hyperplastic versus hypertrophic catch-up fat. Diabetes. 2009;58(5):1037-1039.
doi pubmed - Maury E, Brichard SM. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol Cell Endocrinol. 2010;314(1):1-16.
doi pubmed - Pang C, Gao Z, Yin J, Zhang J, Jia W, Ye J. Macrophage infiltration into adipose tissue may promote angiogenesis for adipose tissue remodeling in obesity. Am J Physiol Endocrinol Metab. 2008;295(2):E313-322.
doi pubmed - Trayhurn P, Wang B, Wood IS. Hypoxia in adipose tissue: a basis for the dysregulation of tissue function in obesity? Br J Nutr. 2008;100(2):227-235.
doi pubmed - Hagström E, Arner P, Ungerstedt U, Bolinder J. Subcutaneous adipose tissue: a source of lactate production after glucose ingestion in humans. Am J Physiol. 1990;258(5 Pt 1):E888-893.
pubmed - Lind L, Lithell H. Decreased peripheral blood flow in the pathogenesis of the metabolic syndrome comprising hypertension, hyperlipidemia, and hyperinsulinemia. Am Heart J. 1993;125(5 Pt 2):1494-1497.
doi - Zimmet P, Boyko EJ, Collier GR, de Courten M. Etiology of the metabolic syndrome: potential role of insulin resistance, leptin resistance, and other players. Ann N Y Acad Sci. 1999;892:25-44.
doi pubmed - Antuna-Puente B, Feve B, Fellahi S, Bastard JP. Adipokines: the missing link between insulin resistance and obesity. Diabetes Metab. 2008;34(1):2-11.
doi pubmed - Alemany M. The defense of adipose tissue against excess substrate-induced hyperthrophia: immune system cell infiltration and arrested metabolic activity. J Clin Endocrinol Metab. 2011;96(1):66-68.
doi pubmed - Umetsu DT, McIntire JJ, Akbari O, Macaubas C, DeKruyff RH. Asthma: an epidemic of dysregulated immunity. Nat Immunol. 2002;3(8):715-720.
doi pubmed - Cohen AD, Sherf M, Vidavsky L, Vardy DA, Shapiro J, Meyerovitch J. Association between psoriasis and the metabolic syndrome. A cross-sectional study. Dermatology. 2008;216(2):152-155.
doi pubmed - Hahn BH, Grossman J, Chen W, McMahon M. The pathogenesis of atherosclerosis in autoimmune rheumatic diseases: roles of inflammation and dyslipidemia. J Autoimmun. 2007;28(2-3):69-75.
doi pubmed - Pasquali R, Vicennati V, Cacciari M, Pagotto U. The hypothalamic-pituitary-adrenal axis activity in obesity and the metabolic syndrome. Ann N Y Acad Sci. 2006;1083:111-128.
- Witchel SF, DeFranco DB. Mechanisms of disease: regulation of glucocorticoid and receptor levels—impact on the metabolic syndrome. Nat Clin Pract Endocrinol Metab. 2006;2(11):621-631.
doi pubmed - Robeva R, Kirilov G, Tomova A, Kumanov P. Melatonin-insulin interactions in patients with metabolic syndrome. J Pineal Res. 2008;44(1):52-56.
pubmed - Muldoon MF, Mackey RH, Sutton-Tyrrell K, Flory JD, Pollock BG, Manuck SB. Lower central serotonergic responsivity is associated with preclinical carotid artery atherosclerosis. Stroke. 2007;38(8):2228-2233.
doi pubmed - Vogelzangs N, Suthers K, Ferrucci L, Simonsick EM, Ble A, Schrager M, Bandinelli S, et al. Hypercortisolemic depression is associated with the metabolic syndrome in late-life. Psychoneuroendocrinology. 2007;32(2):151-159.
doi pubmed - Troxel WM, Buysse DJ, Matthews KA, Kip KE, Strollo PJ, Hall M, Drumheller O, et al. Sleep symptoms predict the development of the metabolic syndrome. Sleep. 2010;33(12):1633-1640.
pubmed - Karmi A, Iozzo P, Viljanen A, Hirvonen J, Fielding BA, Virtanen K, Oikonen V, et al. Increased brain fatty acid uptake in metabolic syndrome. Diabetes. 2010;59(9):2171-2177.
doi pubmed - Coughlin SR, Mawdsley L, Mugarza JA, Calverley PM, Wilding JP. Obstructive sleep apnoea is independently associated with an increased prevalence of metabolic syndrome. Eur Heart J. 2004;25(9):735-741.
doi pubmed - Chin K, Oga T, Takahashi K, Takegami M, Nakayama-Ashida Y, Wakamura T, Sumi K, et al. Associations between obstructive sleep apnea, metabolic syndrome, and sleep duration, as measured with an actigraph, in an urban male working population in Japan. Sleep. 2010;33(1):89-95.
pubmed - Nichols GA, Reinier K, Chugh SS. Independent contribution of diabetes to increased prevalence and incidence of atrial fibrillation. Diabetes Care. 2009;32(10):1851-1856.
doi pubmed - Dela Cruz CS, Matthay RA. Role of obesity in cardiomyopathy and pulmonary hypertension. Clin Chest Med. 2009;30(3):509-523, ix.
doi pubmed - DiGirolamo M. Cellular, metabolic, and clinical consequences of adipose mass enlargement in obesity. Nutrition. 1991;7(4):287-289.
pubmed - Hansen BC, Lewis AJ. Effects of dietary protein concentration (corn:soybean meal ratio) on the performance and carcass characteristics of growing boars, barrows, and gilts: mathematical descriptions. J Anim Sci. 1993;71(8):2122-2132.
pubmed - Calder PC. Branched-chain amino acids and immunity. J Nutr. 2006;136(1 Suppl):288S-293S.
pubmed - Barber T, Viña JR, Viña J, Cabo J. Decreased urea synthesis in cafeteria-diet-induced obesity in the rat. Biochem J. 1985;230(3):675-681.
pubmed - Green LC, Ruiz de Luzuriaga K, Wagner DA, Rand W, Istfan N, Young VR, Tannenbaum SR. Nitrate biosynthesis in man. Proc Natl Acad Sci U S A. 1981;78(12):7764-7768.
doi pubmed - Costa G, Ullrich L, Kantor F, Holland JF. Production of elemental nitrogen by certain mammals including man. Nature. 1968;218(5141):546-551.
doi pubmed - Rees DD, Palmer RM, Moncada S. Role of endothelium-derived nitric oxide in the regulation of blood pressure. Proc Natl Acad Sci U S A. 1989;86(9):3375-3378.
doi pubmed - Frisbee JC. Vascular adrenergic tone and structural narrowing constrain reactive hyperemia in skeletal muscle of obese Zucker rats. Am J Physiol Heart Circ Physiol. 2006;290(5):H2066-2074.
doi pubmed - Muller-Wieland D, Kotzka J, Knebel B, Krone W. Metabolic syndrome and hypertension: pathophysiology and molecular basis of insulin resistance. Basic Res Cardiol. 1998;93 Suppl 2:131-134.
doi pubmed - Sivitz WI, Wayson SM, Bayless ML, Sinkey CA, Haynes WG. Obesity impairs vascular relaxation in human subjects: hyperglycemia exaggerates adrenergic vasoconstriction arterial dysfunction in obesity and diabetes. J Diabetes Complications. 2007;21(3):149-157.
doi pubmed - Frohlich ED, Messerli FH, Reisin E, Dunn FG. The problem of obesity and hypertension. Hypertension. 1983;5(5 Pt 2):III71-78.
pubmed - Weidmann P, de Courten M, Boehlen L, Shaw S. The pathogenesis of hypertension in obese subjects. Drugs. 1993;46 Suppl 2:197-208; discussion 208-199.
- Barton M, Carmona R, Ortmann J, Krieger JE, Traupe T. Obesity-associated activation of angiotensin and endothelin in the cardiovascular system. Int J Biochem Cell Biol. 2003;35(6):826-837.
doi - Cimen MY. Free radical metabolism in human erythrocytes. Clin Chim Acta. 2008;390(1-2):1-11.
doi pubmed - Kennedy L, Lyons TJ. Non-enzymatic glycosylation. Br Med Bull. 1989;45(1):174-190.
pubmed - Cicco G, Pirrelli A. Red blood cell (RBC) deformability, RBC aggregability and tissue oxygenation in hypertension. Clin Hemorheol Microcirc. 1999;21(3-4):169-177.
pubmed - Kuwahira I, Gonzalez NC, Heisler N, Piiper J. Changes in regional blood flow distribution and oxygen supply during hypoxia in conscious rats. J Appl Physiol. 1993;74(1):211-214.
pubmed - Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140(6):900-917.
doi pubmed - Sanz Y, Santacruz A, Gauffin P. Gut microbiota in obesity and metabolic disorders. Proc Nutr Soc. 2010;69(3):434-441.
doi pubmed - Creely SJ, McTernan PG, Kusminski CM, Fisher M, Da Silva NF, Khanolkar M, Evans M, et al. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am J Physiol Endocrinol Metab. 2007;292(3):E740-747.
doi pubmed - Manco M, Putignani L, Bottazzo GF. Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr Rev. 2010;31(6):817-844.
doi pubmed - Gould DC, Amoroso P, Kirby RS. Hypoandrogen-metabolic syndrome: a significant issue for men's health. BJU Int. 2006;98(3):494-496.
doi pubmed - Singh SK, Goyal R, Pratyush DD. Is hypoandrogenemia a component of metabolic syndrome in males? Exp Clin Endocrinol Diabetes. 2011;119(1):30-35.
doi pubmed - Kumar A, Woods KS, Bartolucci AA, Azziz R. Prevalence of adrenal androgen excess in patients with the polycystic ovary syndrome (PCOS). Clin Endocrinol (Oxf). 2005;62(6):644-649.
doi pubmed - Charlton M, Angulo P, Chalasani N, Merriman R, Viker K, Charatcharoenwitthaya P, Sanderson S, et al. Low circulating levels of dehydroepiandrosterone in histologically advanced nonalcoholic fatty liver disease. Hepatology. 2008;47(2):484-492.
doi pubmed - Vegeto E, Benedusi V, Maggi A. Estrogen anti-inflammatory activity in brain: a therapeutic opportunity for menopause and neurodegenerative diseases. Front Neuroendocrinol. 2008;29(4):507-519.
doi pubmed - Prokai L, Prokai-Tatrai K, Perjesi P, Simpkins JW. Mechanistic insights into the direct antioxidant effects of estrogens. Drug Develop Res 2005;66:118-125.
doi - Matthews CE, Sui X, LaMonte MJ, Adams SA, Hebert JR, Blair SN. Metabolic syndrome and risk of death from cancers of the digestive system. Metabolism. 2010;59(8):1231-1239.
doi pubmed - Killinger DW, Strutt BJ, Roncari DA, Khalil MW. Estrone formation from dehydroepiandrosterone in cultured human breast adipose stromal cells. J Steroid Biochem Mol Biol. 1995;52(2):195-201.
doi
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.