









| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website http://www.jofem.org |
Case Report
Volume 6, Number 2, April 2016, pages 64-70
Clinical and Genetic Study of Pseudohypoparathyroidism Type 1b in Hong Kong Chinese
Ho-Ming Luk
Clinical Genetic Service, Department of Health, 3/F Cheung Sha Wan Jockey Club Clinic, 2 Kwong Lee Road, Shamshuipo, Kowloon, Hong Kong SAR, China
Manuscript accepted for publication March 30, 2016
Short title: Pseudohypoparathyroidism Type 1b
doi: http://dx.doi.org/10.14740/jem337w
| Abstract | ▴Top |
Pseudohypoparathyroidism type 1b (PHP-1b) is a rare congenital imprinting disease characterized by parathyroid hormone resistance, normal neurodevelopment and no feature of Albright hereditary osteodystrophy. It is caused by genetic and epigenetic defect of GNAS locus located at chromosome 20q13.32 region. We here have reported a case series of three cases of PHP-1b in Hong Kong Chinese. Two were sporadic and one was autosomal dominant (AD) subtype. All of them were identified by methylation specific-multiplex ligation dependent probe amplification method. The AD PHP-1b case had typical 3-kb STX16 gene deletion. There was no paternal uniparental disomy chromosome 20 for those sporadic cases. Although PHP-1b had been well studied in western population, it is rarely reported in Chinese. This study is the first comprehensive study of PHP-1b for Hong Kong Chinese in the literature. With more awareness and better understanding of PHP-1b, it will result in judicious use of genetic testing and better medical management of this orphan disease.
Keywords: Pseudohypoparathyroidism type 1b; Chinese; Genetics
| Introduction | ▴Top |
Pseudohypoparathyroidism type 1b (PHP-1b) is a congenital imprinting disease that caused by genetic and epigenetic defect of GNAS locus located at chromosome 20q13.32 region. It is characterized by renal resistance to parathyroid hormone (PTH) and occasionally with mild resistance to thyroid stimulating hormone. Typically, PHP-1b patients do not have features of Albright hereditary osteodystrophy (AHO), though obesity, short stature and subtle bony abnormalities have been reported [1].
The GNAS complex contains at least four distinct differentially methylated regions (DMRs). Through parent-of-origin effect with differential methylation on its different promoters, the GNAS locus would give rise to several transcripts, including alpha-subunit of the heterotrimeric stimulatory G protein α (Gαs), the Gαs extra-large variant (XLαs), neuroendocrine protein 55 (NESP55), untranslated exon A/B (exon 1A) and antisense transcript (AS) [2]. The NESP55 is maternal derived while the GNAS XLαs, AS and A/B transcripts are exclusively paternal derived [2]. Consistent with this imprinted expression, the promoters of these genes are silenced through methylation. Therefore, NESP55 should be methylated at paternal allele and the GNAS XLαs, AS and A/B should be methylated at maternal allele in normal individual.
By different mechanism, PHP-1b disease is further divided into autosomal dominant (AD) and sporadic subtypes. In AD PHP-1b, the loss of imprinting at exon A/B DMR is due to microdeletion at the STX16 gene that disrupts its cis-acting control element on Gαs gene [2]. On the other hand, the sporadic PHP-1b has epigenetic abnormalities at multiple DMRs, but without copy number changes in cis- or trans-acting elements [3, 4].
In Hong Kong, sporadic PHP-1b has been reported in the literature [5]; however, there is no comprehensive clinical and molecular study of PHP-1b in tertiary wide basis. Here we have reported a series of three additional cases of PHP-1b in Hong Kong Chinese.
| Case Reports | ▴Top |
Case 1
A 12-year-old girl was referred from private hospital to regional pediatric department for suspected prolonged QTc syndrome and complex partial seizure. She was the first child of non-consanguineous Chinese couple, born at full term via elective lower segment cesarean section. The perinatal history was unremarkable and she enjoyed good past health before admission. Her developmental milestones were normal. She complained of recurrent episodes of facial automatism, eye staring, drooling of saliva and transient unresponsiveness that last for few minutes. All episodes were preceded by abnormal aura. There was no recent febrile illness. Physical examination showed no feature of AHO with body height of 142 cm (-1 SD). Investigation on admission showed hypocalcemia with ionized calcium of 0.84 mmol/L (normal range (NR): 1.1 - 1.35 mmol/L) and hyperphosphatemia at 2.79 mmol/L (NR: 0.72 - 1.43 mmol/L). The renal function and magnesium level were normal. ECG showed prolonged QTc interval of 492 ms at heart rate of 80/min (N: < 440 ms). Intravenous calcium supplement was given to correct the symptomatic hypocalcemia. The QTc interval was normalized and complex partial seizure was stopped afterwards. Further investigations showed elevated PTH at 50.4 pmol/L (NR: 1.6 - 6.9 pmol/L) and moderate vitamin D deficiency with total 25OH vitamin D level of 28 nmol/L (NR: 50 - 220 nmol/L). Thyroid function was normal. Computerized tomography of brain showed no abnormal calcification. She was diagnosed as PHP and put on vitamin D and calcium supplement. She was then regularly followed up in endocrine clinic. Latest calcium and urine calcium/creatinine ratio were normal. Regular ultrasound kidney showed no nephrocalcinosis. The family history was negative for endocrine or developmental problem.
Case 2
An 11-year-old boy was admitted to accident and emergency department for carpopedal spasm. He was the first child of non-consanguineous Chinese couple, born at full term via normal vaginal delivery. The perinatal history was unremarkable. He had history of attention deficient hyperactivity disease and atypical absence seizure that were managed by private neurologist since 10 years old. On admission, the ionized calcium was 0.66 mmol/L (NR: 1.1 - 1.35 mmol/L) and phosphate level was 3.23 mmol/L (NR: 0.72 - 1.43 mmol/L). The renal function and magnesium level were normal. Physical examination showed no feature of AHO with body height of 145 cm (-0.5 SD). There was carpopedal spasm and positive Chvostek’s sign, which have been aborted by correction of hypocalcemia. Further investigations showed elevated PTH at 30.3 pmol/L (NR: 1.6 - 6.9 pmol/L) and mild vitamin D deficiency with total 25OH vitamin D level of 39 nmol/L (NR: 50 - 220 nmol/L). Thyroid function was normal. Computerized tomography of brain showed symmetrical calcification of basal ganglia, and patchy calcification at subcortical U-fibers at bilateral frontal and right parietal region. X-ray hand showed no shortened fourth and fifth metacarpal bone. He was diagnosed as PHP. Vitamin D and calcium supplement was given regularly. The family history was non-contributory. He was then regularly followed up in pediatric unit. There was no recurrence of atypical absence seizure and carpopedal spasm afterwards.
Case 3
A 67-year-old lady was referred to endocrine outpatient clinic for persistent limb numbness. Baseline investigation showed hypocalcemia of 1.46 mmol/L (NR: 2.12 - 2.64 mmol/L) and hyperphosphatemia of 1.93 mmol/L (NR: 1.05 - 1.80 mmol/L). PTH was elevated at 22 pmol/L (NR: 1.6 - 6.9 pmol/L) and mild vitamin D deficiency with total 25OH vitamin D level of 27 nmol/L (NR: 50 - 220 nmol/L). The renal function, alkaline phosphatase, magnesium and thyroid function were normal. Physical examination showed she had body height of 148 cm (-1.5 SD) with mild bilateral shortening of fourth metacarpal bone. However, there was no other evidence of AHO. There was no evidence of other hormone resistance and she had normal intelligence. The family history was negative for endocrine or developmental problem. Based on the biochemical abnormalities, PHP was diagnosed. She was put on calcium together with vitamin D supplement and was regularly followed up in endocrine clinic.
Molecular diagnostic algorithm
Based on hypocalcemia, hyperphosphatemia and elevated PTH, the diagnosis of PHP was substantiated. Without clinical features of AHO and normal intellectual development, the most likely diagnosis was PHP-1b. Therefore, the methylation status of GNAS DMRs together with the copy number change at GNAS DMR and STX16 gene should firstly evaluated by methylation specific-multiplex ligation dependent probe amplification assay (MS-MLPA) by using SALSA MLPA ME031-A2 GNAS kit from MRC-Holland (Amsterdam, The Netherlands) under the manufacturer’s instructions. Paternal uniparental disomy at chromosome 20 (upd(20)pat) was excluded in sporadic PHP-1b by using single nucleotide polymorphism (SNP) array (Agilent SurePrint G3 Human CGH + SNP Microarray 4x180K). Direct sequencing of GNAS gene was also performed in all these patients. Informed consent was obtained prior to the genetic studies.
Results
Total three patients with PHP-1b were reported in this case series. There were two sporadic PHP-1b and one autosomal dominant PHP-1b cases. The MS-MLPA study results are shown in Figure 1. The biochemical abnormalities and genetic alternations are summarized in Table 1. There was no GNAS mutation being detected by sequencing method in these patients. And no upd(20)pat was being detected in those sporadic cases (Fig. 2).
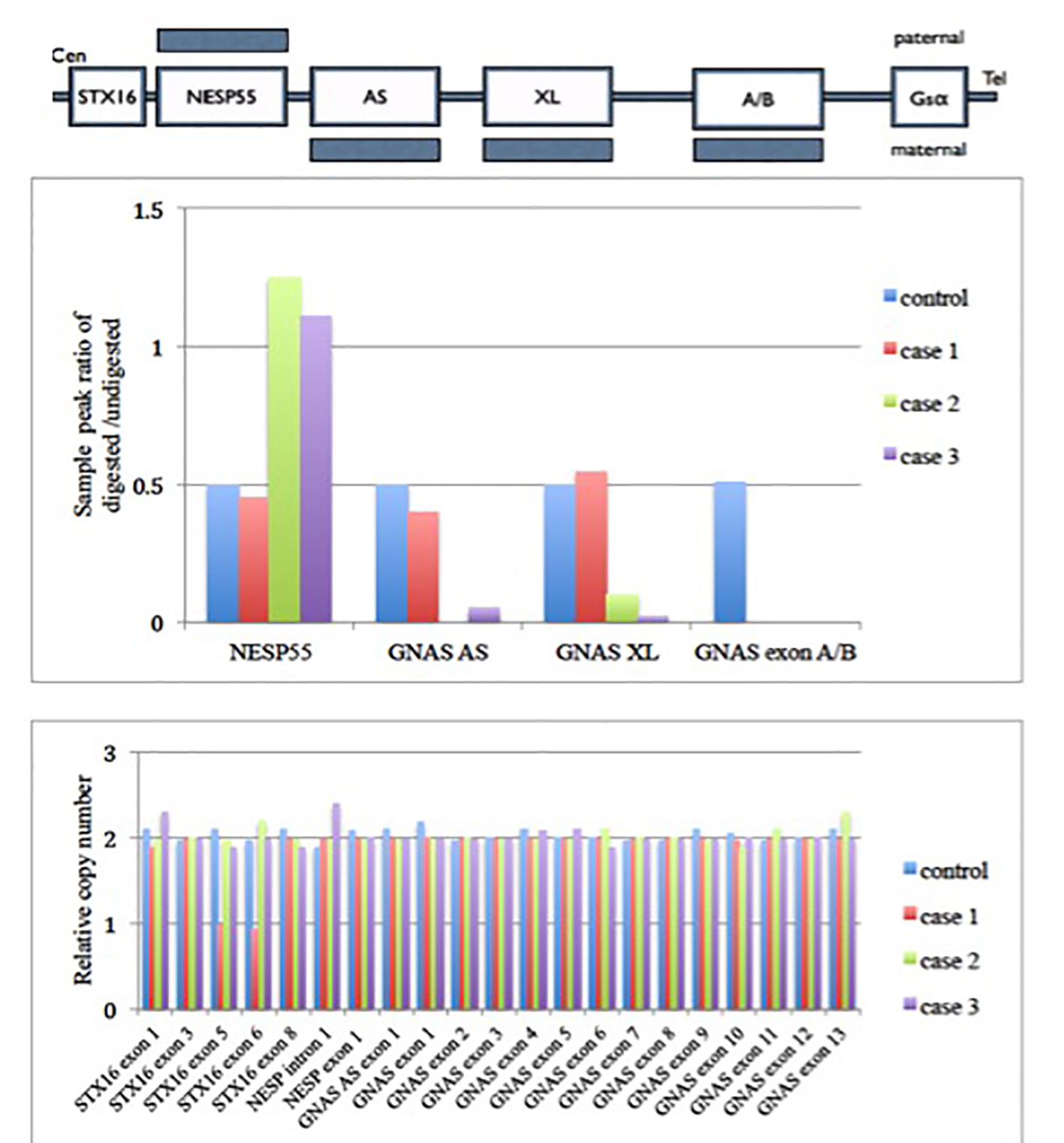 Click for large image | Figure 1. MS-MLPA (GNAS locus) results of these three cases. Upper panel shows a schematic representation of the GNAS locus region. Shaded bars represent the methylation state. Middle panel shows the methylation status of each differential methylation regions (DMRs) at the GNAS locus. The y axis is the ratio of digested to undigested signal change by restriction endonuclease enzyme over GNAS locus. In normal condition, the ratio should be 0.5. Value greater than 0.5 means hypermethylation and less than 0.5 means hypomethylation change over that region. There was hypomethylation at GNAS exon A/B in case 1 patient. There were hypermethylation at the NESP55 and hypomethylation at GNAS AS, GNAS XL and GNAS exon A/B of the patient as compared with control in cases 2 and 3 patients. Lower panel shows the relative copy number change in different exons of GNAS locus. It shows there is deletion of exon 5 and 6 in STX16 gene for case 1. There was no copy number change in GNAS locus among case 2 and 3. Gαs: α subunit of the stimulatory G protein; XLαs: Gαs extra-large variant; NESP55: neuroendocrine protein 55; A/B: untranslated exon A/B; AS: antisense transcript. |
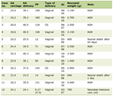 Click to view | Table 1. Summary of the Clinical, Biochemical and Genetic Abnormalities for All PHP-1b Patients in This Study |
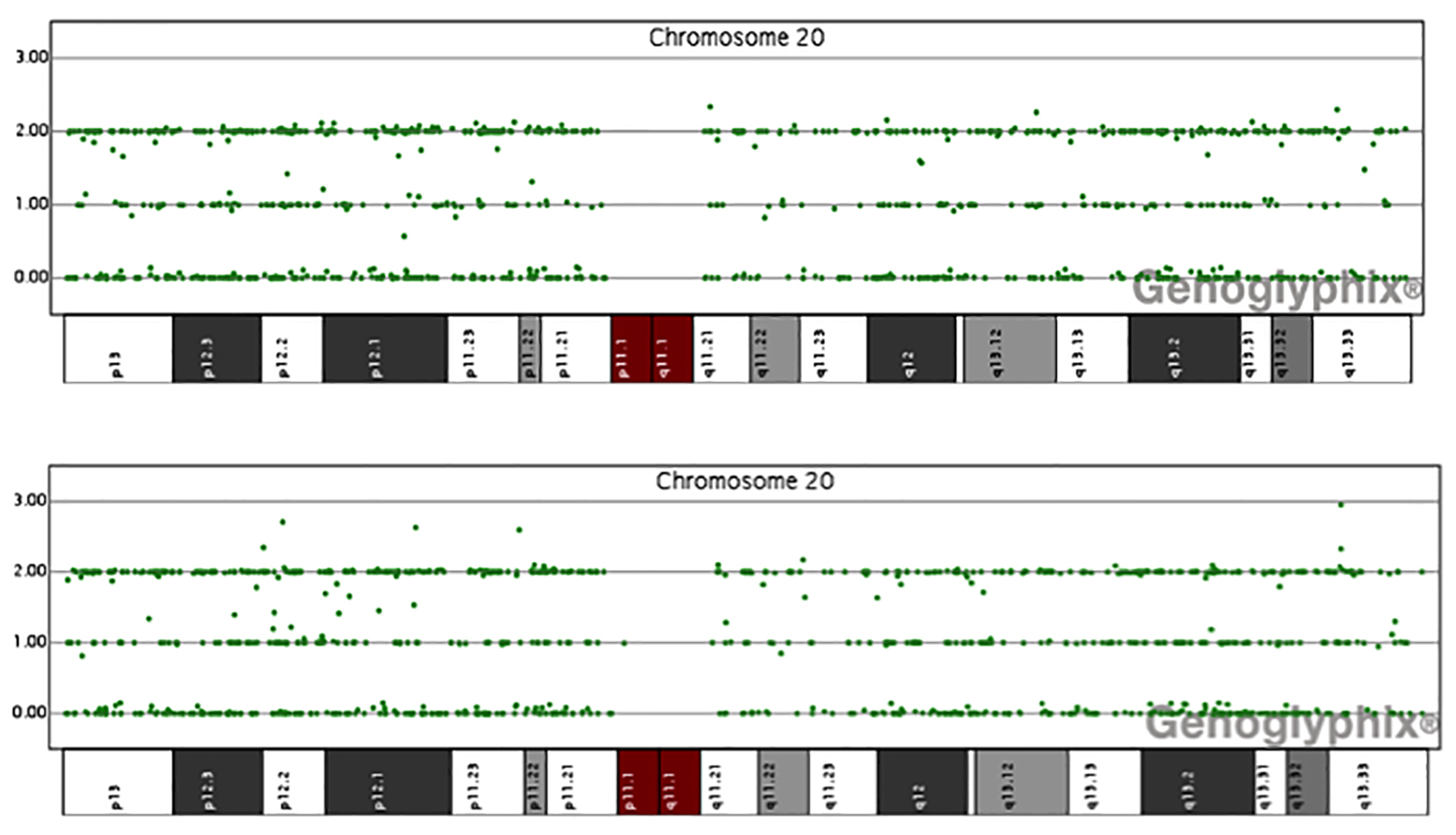 Click for large image | Figure 2. Data of single nucleotide polymorphism (SNP) array of chromosome 20 for patients 2 and 3. The y axis is the number of uncut alleles and x axis represents different region of chromosome 20. In normal condition, there are three lines of signals. In uniparental disomy, the central line signal will lose. Here it shows no evidence of paternal uniparental disomy of chromosome 20 in all our sporadic PHP-1b patients. |
| Discussion | ▴Top |
This is the first comprehensive clinical and genetic case series of PHP-1b patients in Hong Kong Chinese. By using the MS-MLPA method, total three cases of PHP-1b were confirmed molecularly, with the first case of Chinese AD PHP-1b being identified that has not been reported in the literature.
In this AD PHP-1b case, there was 3-kb deletion in the STX16 gene being detected. This deletion encompassed exon 4 to 6 of STX16 gene [6] and was the most frequent genetic mechanism for AD PHP-1b [7-9]. The frequent recurrence was due to the homologous recombination between the repeated sequences in intron 3 and 7 which flanked the deleted region. Apart from 3-kb deletion, maternally inherited microdeletion in other regions within STX16, NESP55 and GNAS AS genes were also reported in AD PHP-1b patients [10-12]. Apart from deletion, maternal duplication of GNAS locus has also been reported to result in PHP-1b phenotype recently [13].
On family cascade screening in this AD PHP-1b family, her mother and younger sister also had the same 3-kb deletion in the STX16 gene. However, methylation study showed there was hypomethylation of GNAS exon A/B in her younger sister but normal methylation status in her mother (Fig. 3). Biochemical screening showed the younger sister had asymptomatic hypocalcemia with elevated PTH, while mother had normal calcium and PTH level. As the maternal grandfather was not available for genetic testing, we postulated that the STX16 gene deletion in her mother should either paternally inherited or happened de novo in the paternal allele that should not have clinical effect on her mother. But once she transmitted this deletion to her daughter, it would exhibit the parent-of-origin imprinting effect and develop PHP-1b disease. This is important for genetic counseling. As the recurrence risk of PHP-1b in maternal carrier was 50%, it has significant implication on her reproductive choice and option in the future. Early identification of those asymptomatic PHP-1b carriers like younger sister of proband is also important as regular anticipatory surveillance and early treatment could reduce the morbidity and even mortality associated with hypocalcemia.
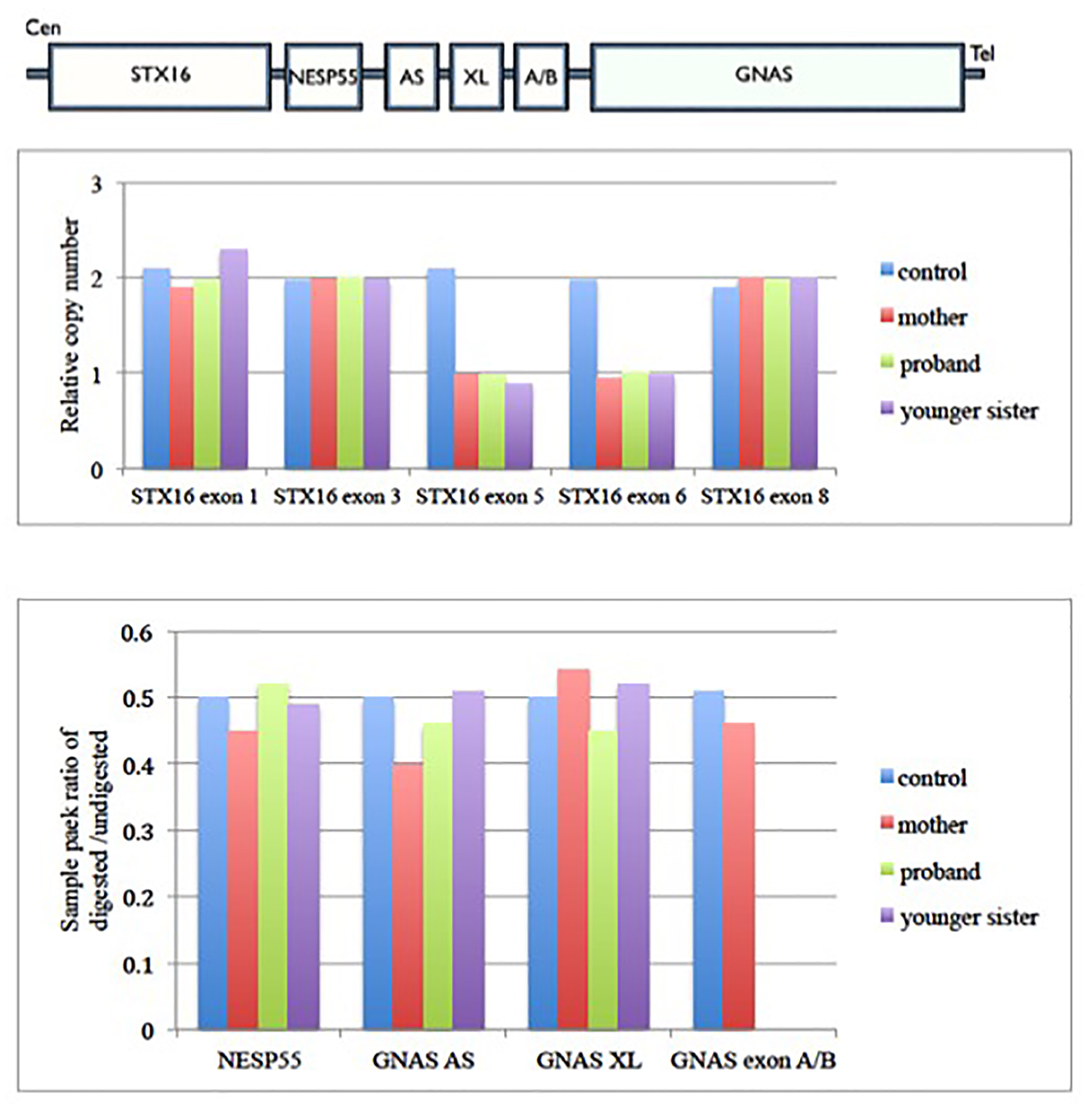 Click for large image | Figure 3. Family cascade screening for case 1 family. Upper panel shows the architecture of GNAS imprinting locus. Middle panel shows the relative copy number change in each exons of STX16 gene in case 1 family. It shows there is deletion of exon 5 and 6 of STX16 gene in proband, her mother and younger sister. Lower panel shows the methylation status of each differential methylation regions (DMRs) at the GNAS locus among this family. The y-axis is the ratio of digested to undigested signal change by restriction endonuclease enzyme over GNAS locus. In normal condition, the ratio should be 0.5. Value greater than 0.5 means hypermethylation and less than 0.5 means hypomethylation change over that region. There was hypomethylation at GNAS exon A/B in proband and her younger sister, but normal methylation status in their mother. Gαs: α subunit of the stimulatory G protein; XLαs: Gαs extra-large variant; NESP55: neuroendocrine protein 55; A/B: untranslated exon A/B; AS: antisense transcript. |
Concerning the genetic testing of PHP-1b, restriction fragment length polymorphism analysis using methylation sensitive enzyme together with Southern blot and bisulfite treated methylation specific PCR were commonly used in the past [14]. However, with the advancement in diagnostic method, MS-MLPA is currently the first line investigation for all suspected PHP-1b patients [6, 15], as it could simultaneously detect the aberrant methylation status of CpG islands and copy number change at the GNAS imprinting locus. Although it is very robust, MS-MLPA method is not without limitation. In MS-MLPA method, only the methylation status of Hhal site in selected CpG islands is being studied. It may not represent the status of entire CpG islands. Moreover, mutation or polymorphism over the probe ligation site will also affect the probe signal intensity [15] that would lead to erroneous results.
Despite recent advancement in understanding the epigenetic defects and PHP-1b, its exact pathogenesis was unknown. The proposed mechanism for AD PHP-1b was STX16 gene deletion which would disrupt the cis-regulatory element of imprinting gene at the GNAS locus [2]. However, the mechanism of epimutation in sporadic PHP-1b that led to abnormal clinical phenotype was still yet to be elucidated. Sporadic PHP-1b accounts for 80-85% of the PHP-1b cases [16]. In the literature, about 2-20% of PHP-1b cases were reported to be associated with upd(20)pat [17-19], thus upd(20)pat should be excluded in all cases of sporadic PHP-1b, either by microsatellites analysis or SNP array study. Since microsatellites analysis is more labor intensive, SNP array might be the first line recommended genetic method to exclude upd(20)pat in sporadic PHP-1b patient. One additional benefit of SNP array is it can measure the size of uniparental disomy. There were reported epigenotype-phenotype correlation between the size of upd(20)pat segment and the clinical features [19, 20]. The longer the segment of upd(20)pat, the earlier the onset of symptoms and the more likely to have overgrowth features like macrosomia and macrocephaly [20]. However, such epigenotype-phenotype correlation could not be demonstrated in non-upd(20)pat related PHP-1b. On the other hand, the degree of methylation aberration was not useful in predicting the severity and type of disease manifestation [21].
With more cases of PHP being studied extensively by molecular technique, it is noted that the GNAS related disease is more genetically heterogeneous and complicated than previously thought. There are molecularly and clinically overlapping between the PHP-1a and PHP-1b diseases [1, 22]. Some PHP-1a patients have epigenetic alternation in GNAS imprinting region and some PHP-1b patients have GNAS gene mutation being reported [23, 24]. Despite this, there was no difference in clinical features and genetic abnormalities between Chinese and non-Chinese PHP patient, therefore based on the current evidence and recommendations [5], we propose the molecular diagnostic algorithm for PHP as Figure 4.
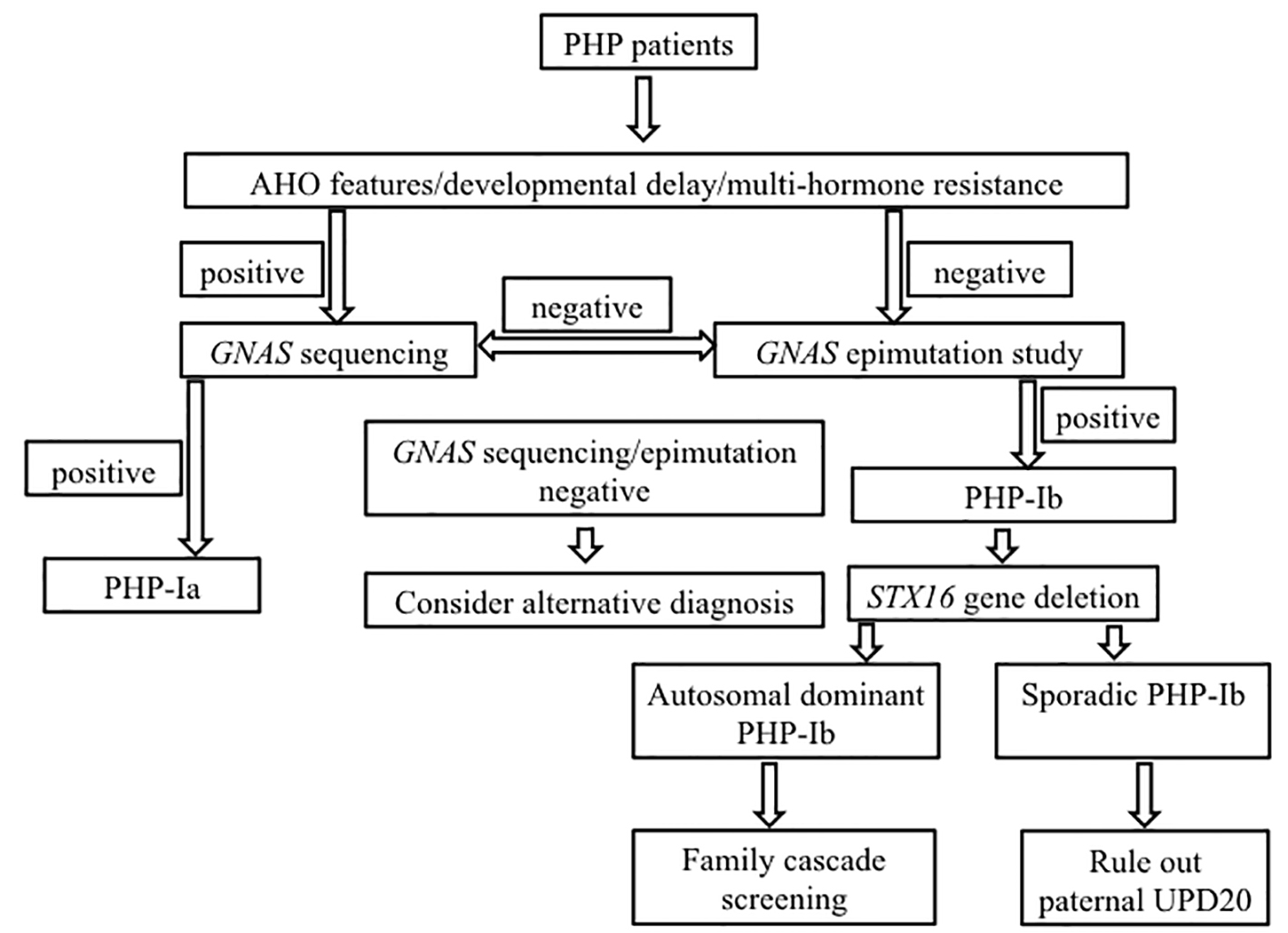 Click for large image | Figure 4. Diagnostic algorithm for molecular genetic testing for patient with PHP. AHO: Albright hereditary osteodystrophy; UPD: uniparental disomy. |
In conclusion, in the presence of hypocalcemia, hyperphosphatemia, elevated PTH, but without intellectual disability or features of AHO, the diagnosis of PHP-1b should be considered. Epimutation study rather than sequencing of the coding region in the GNAS gene should be performed as the first line genetic investigation. Although there is no difference in clinical manifestations between AD PHP-1b and sporadic PHP-1b, their differentiation by genetic testing is of paramount importance for genetic counseling, reproductive choice option and family cascade screening. All cases of PHP-1b should be managed by multidisciplinary teams which included clinical geneticist, so as to provide the best quality of medical care and treatment.
Conflict of Interest
The authors declare that they have no conflict of interest.
| References | ▴Top |
- de Nanclares GP, Fernandez-Rebollo E, Santin I, Garcia-Cuartero B, Gaztambide S, Menendez E, Morales MJ, et al. Epigenetic defects of GNAS in patients with pseudohypoparathyroidism and mild features of Albright's hereditary osteodystrophy. J Clin Endocrinol Metab. 2007;92(6):2370-2373.
doi pubmed - Bastepe M. The GNAS Locus: Quintessential Complex Gene Encoding Gsalpha, XLalphas, and other Imprinted Transcripts. Curr Genomics. 2007;8(6):398-414.
doi pubmed - Bastepe M, Pincus JE, Sugimoto T, Tojo K, Kanatani M, Azuma Y, Kruse K, et al. Positional dissociation between the genetic mutation responsible for pseudohypoparathyroidism type Ib and the associated methylation defect at exon A/B: evidence for a long-range regulatory element within the imprinted GNAS1 locus. Hum Mol Genet. 2001;10(12):1231-1241.
doi pubmed - Liu J, Litman D, Rosenberg MJ, Yu S, Biesecker LG, Weinstein LS. A GNAS1 imprinting defect in pseudohypoparathyroidism type IB. J Clin Invest. 2000;106(9):1167-1174.
doi pubmed - Luk HM, Lo IFM, Tong TMF, Lai KKS, Lam STS. Pseudohypoparathyroidism Type 1b: First Case Report in Chinese and Literature Review. HK J Paediatr (New Series). 2015;20:32-36.
- Alsum Z, Abu Safieh L, Nygren AO, Al-Hamed MA, Alkuraya FS. Methylation-specific multiplex-ligation-dependent probe amplification as a rapid molecular diagnostic tool for pseudohypoparathyroidism type 1b. Genet Test Mol Biomarkers. 2010;14(1):135-139.
doi pubmed - Bastepe M, Frohlich LF, Hendy GN, Indridason OS, Josse RG, Koshiyama H, Korkko J, et al. Autosomal dominant pseudohypoparathyroidism type Ib is associated with a heterozygous microdeletion that likely disrupts a putative imprinting control element of GNAS. J Clin Invest. 2003;112(8):1255-1263.
doi pubmed - Linglart A, Bastepe M, Juppner H. Similar clinical and laboratory findings in patients with symptomatic autosomal dominant and sporadic pseudohypoparathyroidism type Ib despite different epigenetic changes at the GNAS locus. Clin Endocrinol (Oxf). 2007;67(6):822-831.
doi pubmed - Liu J, Nealon JG, Weinstein LS. Distinct patterns of abnormal GNAS imprinting in familial and sporadic pseudohypoparathyroidism type IB. Hum Mol Genet. 2005;14(1):95-102.
doi pubmed - Richard N, Abeguile G, Coudray N, Mittre H, Gruchy N, Andrieux J, Cathebras P, et al. A new deletion ablating NESP55 causes loss of maternal imprint of A/B GNAS and autosomal dominant pseudohypoparathyroidism type Ib. J Clin Endocrinol Metab. 2012;97(5):E863-867.
doi pubmed - Bastepe M, Frohlich LF, Linglart A, Abu-Zahra HS, Tojo K, Ward LM, Juppner H. Deletion of the NESP55 differentially methylated region causes loss of maternal GNAS imprints and pseudohypoparathyroidism type Ib. Nat Genet. 2005;37(1):25-27.
pubmed - Chillambhi S, Turan S, Hwang DY, Chen HC, Juppner H, Bastepe M. Deletion of the noncoding GNAS antisense transcript causes pseudohypoparathyroidism type Ib and biparental defects of GNAS methylation in cis. J Clin Endocrinol Metab. 2010;95(8):3993-4002.
doi pubmed - Perez-Nanclares G, Velayos T, Vela A, Munoz-Torres M, Castano L. Pseudohypoparathyroidism type Ib associated with novel duplications in the GNAS locus. PLoS One. 2015;10(2):e0117691.
doi pubmed - Krueger F, Kreck B, Franke A, Andrews SR. DNA methylome analysis using short bisulfite sequencing data. Nat Methods. 2012;9(2):145-151.
doi pubmed - Yuno A, Usui T, Yambe Y, Higashi K, Ugi S, Shinoda J, Mashio Y, et al. Genetic and epigenetic states of the GNAS complex in pseudohypoparathyroidism type Ib using methylation-specific multiplex ligation-dependent probe amplification assay. Eur J Endocrinol. 2013;168(2):169-175.
doi pubmed - Maupetit-Mehouas S, Azzi S, Steunou V, Sakakini N, Silve C, Reynes C, Perez de Nanclares G, et al. Simultaneous hyper- and hypomethylation at imprinted loci in a subset of patients with GNAS epimutations underlies a complex and different mechanism of multilocus methylation defect in pseudohypoparathyroidism type 1b. Hum Mutat. 2013;34(8):1172-1180.
doi pubmed - Bastepe M, Lane AH, Juppner H. Paternal uniparental isodisomy of chromosome 20q - and the resulting changes in GNAS1 methylation - as a plausible cause of pseudohypoparathyroidism. Am J Hum Genet. 2001;68(5):1283-1289.
doi pubmed - Bastepe M, Altug-Teber O, Agarwal C, Oberfield SE, Bonin M, Juppner H. Paternal uniparental isodisomy of the entire chromosome 20 as a molecular cause of pseudohypoparathyroidism type Ib (PHP-Ib). Bone. 2011;48(3):659-662.
doi pubmed - Fernandez-Rebollo E, Lecumberri B, Garin I, Arroyo J, Bernal-Chico A, Goni F, Orduna R, et al. New mechanisms involved in paternal 20q disomy associated with pseudohypoparathyroidism. Eur J Endocrinol. 2010;163(6):953-962.
doi pubmed - Dixit A, Chandler KE, Lever M, Poole RL, Bullman H, Mughal MZ, et al. Pseudohypoparathyroidism type 1b due to paternal uniparental disomy of chromosome 20q. J Clin Endocrinol Metab. 2013;98:103-108.
doi pubmed - Elli FM, de Sanctis L, Bollati V, Tarantini L, Filopanti M, Barbieri AM, Peverelli E, et al. Quantitative analysis of methylation defects and correlation with clinical characteristics in patients with pseudohypoparathyroidism type I and GNAS epigenetic alterations. J Clin Endocrinol Metab. 2014;99(3):E508-517.
pubmed - Mantovani G, de Sanctis L, Barbieri AM, Elli FM, Bollati V, Vaira V, Labarile P, et al. Pseudohypoparathyroidism and GNAS epigenetic defects: clinical evaluation of albright hereditary osteodystrophy and molecular analysis in 40 patients. J Clin Endocrinol Metab. 2010;95(2):651-658.
doi pubmed - Mariot V, Maupetit-Mehouas S, Sinding C, Kottler ML, Linglart A. A maternal epimutation of GNAS leads to Albright osteodystrophy and parathyroid hormone resistance. J Clin Endocrinol Metab. 2008;93(3):661-665.
doi pubmed - Fernandez-Rebollo E, Garcia-Cuartero B, Garin I, Largo C, Martinez F, Garcia-Lacalle C, Castano L, et al. Intragenic GNAS deletion involving exon A/B in pseudohypoparathyroidism type 1A resulting in an apparent loss of exon A/B methylation: potential for misdiagnosis of pseudohypoparathyroidism type 1B. J Clin Endocrinol Metab. 2010;95(2):765-771.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.