









| Journal of Endocrinology and Metabolism, ISSN 1923-2861 print, 1923-287X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Endocrinol Metab and Elmer Press Inc |
| Journal website http://www.jofem.org |
Case Report
Volume 5, Number 5, October 2015, pages 299-303
Ipilimumab-Induced Hypophysitis May Not Affect All Pituitary Cell Lines: A Case Report
Anupam Kotwala, d, Sarika Raob, Richard Allen Haasc
aDepartment of Medicine, University of Massachusetts Medical School, 55 Lake Avenue North, Room H6-531, Worcester, MA 01655, USA
bDepartment of Endocrine Neoplasia and Hormonal Disorders, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA
cDivision of Endocrinology, Diabetes and Metabolism, Department of Medicine, University of Massachusetts Medical School, 55 Lake Avenue North, Worcester, MA 01655, USA
dCorresponding Author: Anupam Kotwal, Department of Medicine, University of Massachusetts Medical School, 55 Lake Avenue North, Room H6-531, Worcester, MA 01655, USA
Manuscript accepted for publication October 26, 2015
Short title: Ipilimumab-Induced Hypophysitis
doi: http://dx.doi.org/10.14740/jem287w
| Abstract | ▴Top |
Hypophysitis, a rare autoimmune condition, has now emerged as an adverse effect of ipilimumab (anti-cytotoxic T lymphocyte antigen 4 (CTLA4) mAb) therapy. The occurrence of ipilimumab-induced hypophysitis (IH) in studies has varied from 0% to 17%. This condition, because of secondary adrenal insufficiency, may be life-threatening if not recognized and managed promptly. Here we present a case of hypophysitis in the setting of ipilimumab therapy for malignant melanoma. The patient initially presented to his oncologist with a headache, diplopia, fatigue, nausea, hot flashes, anorexia and decreased libido shortly following the third cycle of ipilimumab. He was found to have pituitary enlargement with heterogeneous enhancement on magnetic resonance imaging (MRI). The initial diagnosis was based on clinical features, MRI findings and laboratory evidence of central hypogonadism and adrenal insufficiency. Other hormone levels were not tested that time and were only tested 5 months later when the patient did not tolerate the discontinuation of glucocorticoids. He continued to require glucocorticoid and testosterone replacement 14 months after the diagnosis. This case demonstrates IH causing anterior hypopituitarism leading to central hypoadrenalism, central hypogonadism, reduced prolactin and possibly central hypothyroidism but preserved somatotroph function. Pituitary antibodies may be a possible method of detection of IH in addition to MRI and hormonal investigations. There is insufficient evidence to support the need for discontinuing ipilimumab or the superiority of initial high dose versus physiologic steroid and hormone replacement in the management of IH. Hypopituitarism due to IH may persist for several months or longer after ipilimumab is discontinued. This underlies the importance of continuous supplementation with all the hormones that are deficient as a consequence of hypopituitarism caused by IH.
Keywords: Ipilimumab; Hypophysitis; Hypopituitarism
| Introduction | ▴Top |
Autoimmune lymphocytic hypophysitis is the most common chronic inflammation to primarily affect the pituitary gland [1]. It has been divided into adenohypophysitis, infundibulohypophysitis, neurohypophysitis or panhypophysitis depending on the anatomical location involved, with adenohypophysitis being the most commonly reported. This rare condition has now emerged as an adverse effect of immune-modulatory therapy for malignancy. Cytotoxic T lymphocyte antigen 4 (CTLA4) is a key immune checkpoint molecule that down regulates T-cell activation and proliferation. It contributes to controlling autoimmunity, and in the presence of cancer, it limits the expansion of tumor-specific effector T cells, favoring cancer immune tolerance. Ipilimumab is an anti-CTLA4 monoclonal antibody which by blocking this molecule leads to enhanced T-cell activation and antitumor effects. It was approved by the US FDA in 2011 for treatment for metastatic or unresectable melanoma. The approved dose is 3 mg/kg administered as an intravenous infusion every 3 weeks for a total of four doses. Survival benefit has been demonstrated with 3 mg/kg [2], but not with lower doses. In some patients, maintenance therapy may continue with additional infusions at longer intervals. Multiple clinical trials have demonstrated improved survival in cancers especially malignant melanoma with ipilimumab therapy [2, 3]. Many of these trials have reported the occurrence of immune-related adverse effects (IRAEs) including endocrinopathies, colitis, dermatitis and hepatitis. Although the most commonly reported endocrinopathy is hypophysitis, rarely thyroiditis and occasionally adrenalitis [4-7] have been reported.
Ipilimumab-induced hypophysitis (IH), because of secondary adrenal insufficiency, may be life-threatening if not recognized and managed promptly [1]. Although a significant number of cases have been previously reported, there still remain major areas of uncertainty regarding the exact mechanism, risk factors, course and prognosis of this condition. Some studies have reported the cumulative dose of ipilimumab to contribute to the development of hypophysitis, whereas others have refuted this claim. The exact antigenic target(s) for this condition is still not elucidated. Studies have reported variable incidence of IH, and the risk factors for development remain largely undefined. Long-term follow-up is available for only a few cases, and it is unclear if the occurrence of IH predicts the length of survival. Here we present a case of hypophysitis in the setting of ipilimumab therapy for malignant melanoma and review the diagnosis and management of this condition.
| Case Report | ▴Top |
A 68-year-old Caucasian gentleman presented to the oncology clinic with complaints of headache, diplopia, fatigue, nausea, hot flashes, anorexia and decreased libido. Four months prior to presentation, he had been diagnosed with melanoma that metastasized to the retro-peritoneum and cerebellum. He underwent resection of the solitary cerebellar metastasis following which ipilimumab was initiated. His symptoms developed shortly after the third cycle of 3 mg/kg ipilimumab therapy for metastatic melanoma. His medical history at the time of diagnosis was significant for total thyroidectomy for multinodular goiter, and he had been on levothyroxine for the past 40 years. Prior to the initiation of ipilimumab, he was noted to be euthyroid on replacement therapy; however, the adrenal function was not tested. When he presented to the oncologist, he was found to have mild proximal muscle weakness. Laboratory workup revealed a low AM cortisol, low total testosterone, low TSH, normal free T4, low total T3 and normal serum electrolytes (Table 1). Magnetic resonance imaging (MRI) of the brain demonstrated pituitary enlargement with heterogeneous enhancement after contrast administration (Fig. 1). This was a new finding as compared to an MRI done 4 months previously. Based on these clinical, laboratory and radiologic findings, he was diagnosed with IH by the oncologist. Prednisone, at a dose of 90 mg (1 mg/kg) daily, and topical testosterone gel were initiated. He also developed dermatitis and colitis that were attributed to being IRAEs; hence he did not receive further ipilimumab therapy. On follow-up MRI, the pituitary enlargement and enhancement resolved within 3 months appearing like an empty sella (Fig. 2). He demonstrated clinical improvement, and the prednisone was gradually tapered to 5 mg daily. The patient had discontinued topical testosterone by himself a month after diagnosis as his hot flashes had resolved.
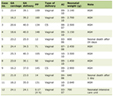 Click to view | Table 1. Laboratory Investigations |
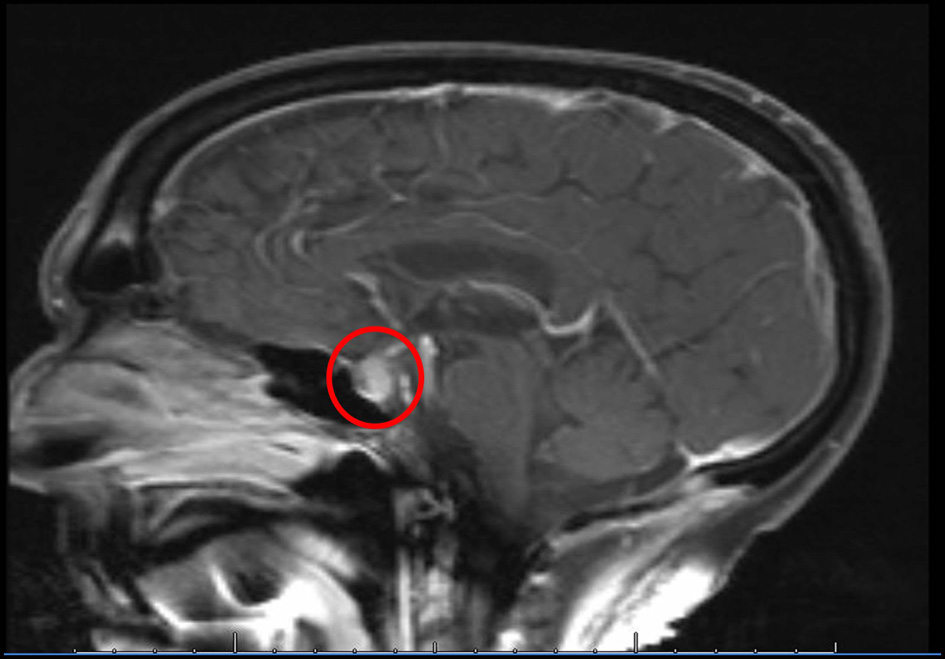 Click for large image | Figure 1. MRI of the brain performed at the time of diagnosis of IH demonstrates soft tissue fullness of the pituitary with heterogeneous enhancement after contrast. |
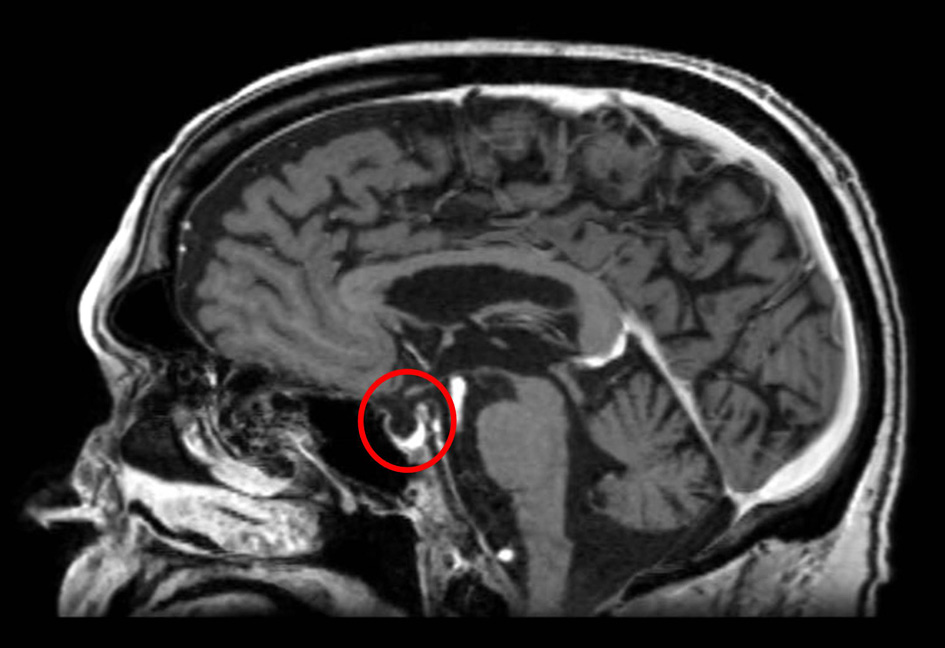 Click for large image | Figure 2. MRI of the brain 3 months later demonstrates resolution of the pituitary enhancement with appearance of an empty sella. |
Approximately 5 months after the diagnosis of IH, prednisone was discontinued and within 1 week his symptoms of fatigue, nausea, anorexia and reduced libido recurred. He was then referred to the endocrinology clinic at which time he was found to have a Cushingoid appearance, mild proximal muscle weakness, and a 14 lb weight gain over the past 5 months. Hormonal investigations revealed a low AM serum cortisol, low ACTH, abnormal ACTH stimulation test, low TSH, normal free T4, low prolactin, low total testosterone, normal IGF-1 and an inappropriately normal LH level. These findings in addition to his symptoms were consistent with persistent anterior hypopituitarism. His testosterone was profoundly low, and he had been off exogenous testosterone replacement for approximately 4 months. Serum and urine osmolality, as well as electrolytes, were within the normal range. Hydrocortisone replacement of 30 mg daily in two divided doses was initiated, and topical testosterone gel was resumed. Levothyroxine dose was decreased from 112 to 100 μg daily. He was soon diagnosed with obstructive sleep apnea, and nocturnal continuous positive airway pressure was initiated, following which his fatigue dramatically improved. Hence, the dose of hydrocortisone was reduced to 20 mg daily in two divided doses. At 8-month follow-up, his symptoms had almost resolved and the testosterone level had improved. At 14-month follow-up, ACTH and AM serum cortisol tested after holding the morning dose of hydrocortisone resulted low, prolactin level was low, TSH was low and total testosterone was slightly lower than that at 8-month follow-up. His only complaint was reduced libido; hence the testosterone dose was increased. The hydrocortisone dose was decreased to 15 mg daily in two divided doses.
| Discussion | ▴Top |
We present a patient with malignant melanoma who developed features of adenohypophysitis after the third cycle of ipilimumab therapy, and presented with pituitary mass effect as well as anterior hypopituitarism. Ipilimumab increases T-cell activity by blocking the inhibitory receptor CTLA4, leading to antitumor effects. However, the same mechanism might cause a spectrum of inflammatory side-effects classified as IRAEs. Autoimmunity has been proposed to be the mechanism responsible for these adverse effects [8, 9]. The occurrence of IH in studies has varied from 0% to 17% [2, 8, 10-15]; however, these studies have used different doses of ipilimumab ranging from 0.3 to 10 mg/kg. In a recent case series, 11% of patients undergoing treatment with ipilimumab developed hypophysitis [5]. Based on a review, there appears to be a dose-dependent increase in the occurrence of IH [10]. At the lower ipilimumab dose (1 - 3 mg/kg), it occurred in 1.8-3.3% of cases [8, 9]. When the dose exceeds 3 mg/kg, the occurrence of IH varies from 4.9 to 17% [13, 14]. Our patient developed symptoms after the third cycle of 3 mg/kg ipilimumab therapy for malignant melanoma. Most but not all cases of IH have been reported to occur after a similar duration of therapy. According to a review, patients who received 3 mg/kg ipilimumab developed symptoms at a median time of 11 weeks (that is, before the fourth dose), suggesting a possible cumulative effect [16]. This has also been supported by another recent review of clinical trials [4]. However, a study in which patients used 10 mg/kg ipilimumab reported that hypophysitis symptoms occurred after the first infusion (4 weeks) in one patient and after the fourth (16 weeks) in another [10], and a recent prospective cohort study did not find the cumulative dose of ipilimumab to be a risk factor for hypophysitis [5].
The clinical presentation of IH relates to pituitary mass effect and hormone deficiencies. Affected individuals may present with nonspecific symptoms such as fatigue, weakness, headache, nausea, visual impairments such as diplopia (less frequent than in classic lymphocytic autoimmune hypophysitis),confusion, memory loss, loss of libido, anorexia, insomnia, hallucinations, temperature intolerance, and subjective sensation of fever and chills [10]. Earlier studies have reported that ACTH and TSH seem to be invariably lost, and most male patients have hypogonadotropic hypogonadism [10, 14, 16]. Faje et al reported all cases with IH to have central hypothyroidism and hypogonadism; however, only approximately 50% of the affected individuals had secondary adrenal insufficiency [5]. The levels of IGF-1 were tested in only six of the 17 affected individuals and found to be low in one individual, and the prolactin level was found to be low in most of the affected individuals [5]. Faje et al did not report any cases of diabetes insipidus due to ipilimumab [5], and we could find only one case in the literature reporting this [10]. The pattern of loss of pituitary function from IH appears to be similar to classic autoimmune hypophysitis but different from other causes of anterior hypopituitarism. In most but not all cases of IH, MRI reveals enlargement of the pituitary gland (up to 60-100% of baseline size), with thickening of the stalk [5, 14]. In the case series by Min et al, only one out of eight patients had typical hypophysitis MRI findings, which resolved after 1 month [15]. The pituitary enlargement preceded the clinical diagnosis of hypophysitis in eight out of 17 IH patients, often by several weeks in the study by Faje et al [5]. The MRI findings in IH have been reported to be lesser in magnitude as compared to classic lymphocytic autoimmune hypophysitis [15]. Our patient was found to have pituitary enlargement with heterogeneous contrast enhancement at the time of initial diagnosis. The initial diagnosis was based on clinical features, MRI findings and laboratory evidence of adrenal insufficiency and hypogonadism by the patient’s oncologist. Other hormone levels were not tested at that time and were only tested later when the patient did not tolerate the discontinuation of glucocorticoids. He then demonstrated central hypoadrenalism and hypogonadotropic hypogonadism but preserved somatotroph function. The decrease in TSH in our patient could be attributed to hypopituitarism causing central hypothyroidism; however, his history of post-surgical hypothyroidism requiring levothyroxine supplementation complicates the picture. Our patient developed a decrease in prolactin similar to some reported cases, whereas others have demonstrated an increase in prolactin likely due to stalk involvement [14]. Serum and urine osmolality, as well as electrolytes, were within the normal range, suggesting that posterior pituitary and renin-angiotensin-aldosterone functions were preserved.
The recommended treatment for most IRAEs secondary to anti-CTLA4 therapy consists of high-dose glucocorticoids (dexamethasone 4 mg every 6 h, prednisolone 45 - 60 mg daily or prednisone 1 mg/kg daily), tapered down gradually over 1 month. In cases of autoimmune hypophysitis, the initial high dose of glucocorticoids should be replaced by a physiologic dose of hydrocortisone of 15 - 20 mg/day in divided doses. In the study by Faje et al, IH patients treated with high-dose glucocorticoids typically received prednisone 60 mg daily, followed by a taper to a physiologic replacement dose over a variable period of time (often 6 - 8 weeks) [5]. Although many patients with IH receive high doses of glucocorticoids, it is not clear whether this practice is necessary or represents an optimal management strategy in this population [4]. It remains to be clarified whether these initial high doses of glucocorticoids are needed, or whether from the time of diagnosis patients should be started on close to a physiologic replacement dose. There is a theoretical concern that high-dose glucocorticoids might hinder the antitumor effect of ipilimumab; however, the available evidence does not support this [12, 14]. Our patient initially received high-dose glucocorticoid, which was then tapered to physiologic dose. The occurrence of other IRAEs may require discontinuation of ipilimumab; however, the patients who continued it in the study by Faje et al showed a similar course of hypopituitarism on follow-up [5]. Based on the available evidence and development of colitis and dermatitis, ipilimumab was discontinued in our patient.
Almost all reported cases of IH experience resolution of acute symptoms a few days after ipilimumab discontinuation and starting high-dose glucocorticoids and other hormone replacement [14]. On follow-up, the pituitary enhancement has been shown to decrease on glucocorticoid replacement at a slower rate in comparison to the clinical response [16]. The MRI findings in our patient resolved 3 months after ipilimumab discontinuation and high-dose glucocorticoid initiation. His symptoms due to pituitary mass effect also resolved and did not recur. Pituitary function may be impaired for a longer period despite high-dose glucocorticoid therapy. Faje et al reported hypopituitarism to be persistent in most cases (13 out of 17) at the time of last follow-up (median length of follow-up 11.5 months), adrenal recovery occurred in one patient, thyroidal recovery was observed in one patient, gonadal function normalized in two patients, and pituitary hormone deficiencies did recover in one patient 3 years after the diagnosis of IH [5]. In the largest clinical trial of 163 patients treated with ipilimumab, regardless of management, all patients with hypophysitis continued to require replacement glucocorticoids with 4 - 26 months follow-up and some continued to require thyroxine and testosterone replacement[14]. In our patient, the discontinuation of glucocorticoid and testosterone caused symptomatic worsening. At that time, he demonstrated central hypoadrenalism, hypogonadotropic hypogonadism in the absence of exogenous testosterone, and decreased prolactin. After re-initiation of slightly higher than physiologic hydrocortisone and testosterone replacement, his clinical status improved. We were able to decrease the hydrocortisone to physiologic dose after treatment of his obstructive sleep apnea that improved his fatigue. Fourteen months after being diagnosed with IH, he has continued to require testosterone and hydrocortisone replacement. This suggests that hypopituitarism due to IH may persist for several months or longer after diagnosis.
Some studies have reported the development of autoimmunity to be associated with increased tumor response [8, 17]. Faje et al reported increased median survival in individuals who developed IH (19.4 months) in comparison to those who did not (8.8 months). In terms of risk factors, IH has demonstrated a higher predilection for males [4, 5] and older individuals [5]. This is different from classic lymphocytic hypophysitis that shows a higher female preponderance [1]. It is unclear why the anterior pituitary is particularly susceptible to ipilimumab therapy. Although polymorphisms in the CTLA4 gene have been suggested to be related to certain autoimmune endocrinopathies such as Grave’s disease and Hashimoto thyroiditis [18, 19], there have been no reports describing the association of such polymorphisms with IH. Iwama et al have demonstrated the development of antibodies against corticotrophs, thyrotrophs and gonadotrophs in patients with IH [20]. This might explain an underlying mechanism for the autoimmune hypophysitis caused by ipilimumab. However, the exact antigenic target(s) for this condition remains to be elucidated. In any patient with malignancy treated with ipilimumab who develops features concerning for hypophysitis, metastasis to the pituitary should be considered, especially if the pituitary enlargement does not respond to glucocorticoid therapy [4].
Conclusions
Autoimmune hypophysitis has emerged as an IRAE of ipilimumab (anti-CTLA4 mAb). The clinical presentation of IH relates to pituitary mass effect and hypopituitarism. It usually affects the anterior pituitary, most commonly involved cell lines being corticotrophs, thyrotrophs and gonadotrophs, with lesser-reported involvement of somatotrophs. Prolactin may be elevated or reduced and posterior pituitary is usually spared. Pituitary antibodies might be a possible method of detection in addition to hormonal and radiological investigations; however, the antigenic target remains to be elucidated. There is insufficient evidence to support the need for discontinuing ipilimumab or the superiority of initial high-dose glucocorticoids versus physiologic hormone replacement in the management of IH. Most clinical and radiological features of IH are consistent with classic lymphocytic hypophysitis; however, the aspects in which these differ are male preponderance, increased incidence in older age and lesser magnitude of MRI findings reported with IH. Pituitary function should be tested before starting ipilimumab, and when IH is suspected, it should be managed promptly as the resulting adrenal insufficiency may be life-threatening. Failure of resolution of pituitary enlargement should raise the concern for metastasis of the primary malignancy to the pituitary. Hypopituitarism due to IH usually persists for several months or longer after diagnosis even though pituitary mass effect resolves after glucocorticoid therapy. This underlies the importance of continuous replacement of the deficient hormones, unless there is evidence of recovery of pituitary function.
Conflict of Interest
The authors have no conflicts of interest.
Abbreviations
CTLA4: cytotoxic T lymphocyte antigen 4; IRAEs: immune-related adverse effects; IH: ipilimumab-induced hypophysitis; MRI: magnetic resonance imaging
| References | ▴Top |
- Caturegli P, Newschaffer C, Olivi A, Pomper MG, Burger PC, Rose NR. Autoimmune hypophysitis. Endocr Rev. 2005;26(5):599-614.
doi pubmed - Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711-723.
doi pubmed - Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, Lebbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517-2526.
doi pubmed - Corsello SM, Barnabei A, Marchetti P, De Vecchis L, Salvatori R, Torino F. Endocrine side effects induced by immune checkpoint inhibitors. J Clin Endocrinol Metab. 2013;98(4):1361-1375.
doi pubmed - Faje AT, Sullivan R, Lawrence D, Tritos NA, Fadden R, Klibanski A, Nachtigall L. Ipilimumab-induced hypophysitis: a detailed longitudinal analysis in a large cohort of patients with metastatic melanoma. J Clin Endocrinol Metab. 2014;99(11):4078-4085.
doi pubmed - Min L, Ibrahim N. Ipilimumab-induced autoimmune adrenalitis. Lancet Diabetes Endocrinol. 2013;1(3):e15.
doi - Min L, Vaidya A, Becker C. Thyroid autoimmunity and ophthalmopathy related to melanoma biological therapy. Eur J Endocrinol. 2011;164(2):303-307.
doi pubmed - Attia P, Phan GQ, Maker AV, Robinson MR, Quezado MM, Yang JC, Sherry RM, et al. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J Clin Oncol. 2005;23(25):6043-6053.
doi pubmed - Yang JC, Hughes M, Kammula U, Royal R, Sherry RM, Topalian SL, Suri KB, et al. Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J Immunother. 2007;30(8):825-830.
doi pubmed - Dillard T, Yedinak CG, Alumkal J, Fleseriu M. Anti-CTLA-4 antibody therapy associated autoimmune hypophysitis: serious immune related adverse events across a spectrum of cancer subtypes. Pituitary. 2010;13(1):29-38.
doi pubmed - Maker AV, Phan GQ, Attia P, Yang JC, Sherry RM, Topalian SL, Kammula US, et al. Tumor regression and autoimmunity in patients treated with cytotoxic T lymphocyte-associated antigen 4 blockade and interleukin 2: a phase I/II study. Ann Surg Oncol. 2005;12(12):1005-1016.
doi pubmed - Weber JS, O'Day S, Urba W, Powderly J, Nichol G, Yellin M, Snively J, et al. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol. 2008;26(36):5950-5956.
doi pubmed - Maker AV, Yang JC, Sherry RM, Topalian SL, Kammula US, Royal RE, Hughes M, et al. Intrapatient dose escalation of anti-CTLA-4 antibody in patients with metastatic melanoma. J Immunother. 2006;29(4):455-463.
doi pubmed - Blansfield JA, Beck KE, Tran K, Yang JC, Hughes MS, Kammula US, Royal RE, et al. Cytotoxic T-lymphocyte-associated antigen-4 blockage can induce autoimmune hypophysitis in patients with metastatic melanoma and renal cancer. J Immunother. 2005;28(6):593-598.
doi pubmed - Min L, Vaidya A, Becker C. Association of ipilimumab therapy for advanced melanoma with secondary adrenal insufficiency: a case series. Endocr Pract. 2012;18(3):351-355.
doi pubmed - Juszczak A, Gupta A, Karavitaki N, Middleton MR, Grossman AB. Ipilimumab: a novel immunomodulating therapy causing autoimmune hypophysitis: a case report and review. Eur J Endocrinol. 2012;167(1):1-5.
doi pubmed - Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, Restifo NP, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U S A. 2003;100(14):8372-8377.
doi pubmed - Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, Rainbow DB, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423(6939):506-511.
doi pubmed - Chistiakov DA, Turakulov RI. CTLA-4 and its role in autoimmune thyroid disease. J Mol Endocrinol. 2003;31(1):21-36.
doi - Iwama S, De Remigis A, Callahan MK, Slovin SF, Wolchok JD, Caturegli P. Pituitary expression of CTLA-4 mediates hypophysitis secondary to administration of CTLA-4 blocking antibody. Sci Transl Med. 2014;6(230):230ra245.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Endocrinology and Metabolism is published by Elmer Press Inc.